
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA CuI/CuIII prototypical organometallic mechanism for the deactivation of an active pincer-like CuI catalyst in Ullmann-type couplings†
Mireia
Rovira
a,
Lucie
Jašíková
b,
Erik
Andris
b,
Ferran
Acuña-Parés
 a,
Marta
Soler
a,
Imma
Güell
a,
Ming-Zheng
Wang
a,
Laura
Gómez
ac,
Josep M.
Luis
a,
Jana
Roithová
*b and
Xavi
Ribas
*a
a,
Marta
Soler
a,
Imma
Güell
a,
Ming-Zheng
Wang
a,
Laura
Gómez
ac,
Josep M.
Luis
a,
Jana
Roithová
*b and
Xavi
Ribas
*a
aInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, E-17003 Girona, Catalonia, Spain. E-mail: xavi.ribas@udg.edu
bDepartment of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic. E-mail: jana.roithova@natur.cuni.cz
cServeis Tècnics de Recerca (STR), Universitat de Girona, Parc Científic i Tecnològic de la UdG, Pic de Peguera 15, E-17003 Girona, Catalonia, Spain
First published on 13th July 2017
Abstract
Unraveling the mechanistic details of copper-catalyzed arylation of nucleophiles (Ullmann-type couplings) is a very challenging task. It is a matter of intense debate whether it is a radical-based process or an organometallic redox-based process. The ancillary ligand choice in Ullmann-type couplings plays a key role in such transformations and can strongly influence the catalytic efficiency as well as the mechanism. Here, we show how a predesigned tridentate pincer-like catalyst undergoes a deactivation pathway through a CuI/CuIII prototypical mechanism as demonstrated by helium-tagging infrared photodissociation (IRPD) spectroscopy and DFT studies, lending a strong support to the existence of an aryl–CuIII species in the Ullmann couplings using this tridentate ligand.
Modern Cu-catalyzed cross-coupling reactions have recently evolved into reliable and efficient methods for the formation of C–C and C-heteroatom bonds, which are present in a large number of natural and pharmaceutical products.1–3 Unravelling the mechanistic details of copper-catalyzed arylation of nucleophiles (Ullmann-type couplings, Scheme 1a) is a very challenging task and a matter of intense debate in order to verify which of the two main mechanistic proposals is taking place: a radical-based or an organometallic-based process.4,5
 | ||
| Scheme 1 (a) Copper-catalyzed Ullmann-type C-heteroatom couplings and (b) the two main mechanistic proposals. | ||
Since the late 1990s, much effort has been devoted to the use of chelating ligands, such as diamines, triamines, amino acids, phenanthroline derivatives and β-diketones, to perform coupling reactions under milder conditions while achieving enhanced yields.3,6,7 The detection of intermediate species after the activation of the aryl halide, which is usually rate-limiting, is very limited and most mechanistic proposals are derived from kinetic and computational studies.8–12 There is an ongoing discussion in the literature concerning the mechanistic pathway for Ullmann condensation reactions. The most invoked mechanisms for Ullmann couplings are based on either one-electron redox processes through radical intermediates that may operate via a CuI/CuII catalytic cycle, or two-electron redox processes via a CuI/CuIII catalytic cycle (Scheme 1b).5 Recently, Peters, Fu and co-workers reported a photoluminescent Cu–carbazolide complex bearing monophosphine ligands for promoting the photoinduced C–N bond-forming reaction.13–15 Upon photoexcitation of the Cu–carbazolide complex, a Cu-containing radical is formed, as detected by EPR spectroscopy. This radical intermediate reacts with aryl halides via Single Electron Transfer (SET) to afford the corresponding C–N coupling product. Therefore, it is likely that several mechanisms might be possible simultaneously and the experimental conditions used are crucial to determine the operative one.
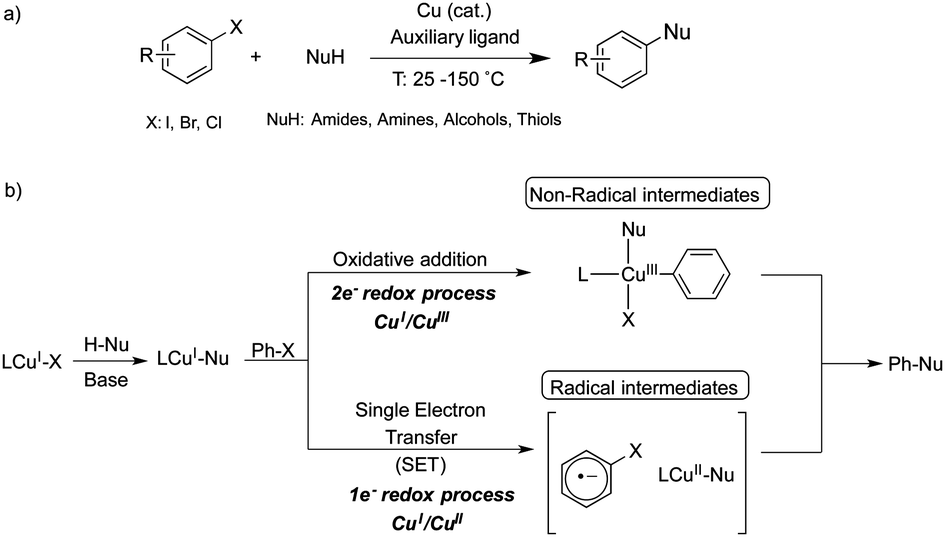
Focusing our attention on the design of appropriate auxiliary ligands for Ullmann couplings under standard thermal conditions, we have previously demonstrated that a CuI/CuIII catalytic cycle is operative in model macrocyclic aryl halide substrates, where active aryl–CuIII–X species have been completely characterized within these systems.5,16,17 Finding inspiration in the macrocyclic model systems, in this work we specifically design auxiliary ligand L3 (Fig. 1) to reproduce the equivalent geometry of the Cu center, so that the stabilization of aryl–CuIII might become possible (Fig. 1).
 | ||
| Fig. 1 Design of the auxiliary ligand L3 used in this work. | ||
In order to gain insight into the plausible operative mechanisms, we initially undertook a Cold-Spray18 MS study using L3 as the auxiliary ligand for the C–O Ullmann-type coupling of iodobenzene and p-methoxyphenol (2) as a model reaction (a standard 24 h reaction afforded a 45% yield of the diarylether product).19 Notably, an intense peak at m/z = 304.0877 was detected in positive ESI mode after 0.5 hour (see the ESI for details of the procedure). At first sight, this peak could correspond to a putative CuIII intermediate species involved in the reaction ([L3′–CuIII–(C6H5)]+), L3′ being the monodeprotonated version of L3 (Fig. S1, ESI†). We found the same analogous peak when using either bromobenzene (m/z = 304.0877), 1-iodo-4-methylbenzene (m/z = 318.1008) or 1-iodo-3,5-dimethylbenzene (332.1167) (Fig. S2–S4, ESI†). Additionally, when using geometrically similar tridentate ligands L4 (2,6-pyridinediyldimethanamine) and L5 (N,N′-diethyl-2,6-bis(aminomethylpyridine)), peaks analogous to 304.0877 were also observed at m/z = 276.0540 and 332.1144, respectively (Fig. S5 and S6, ESI†).
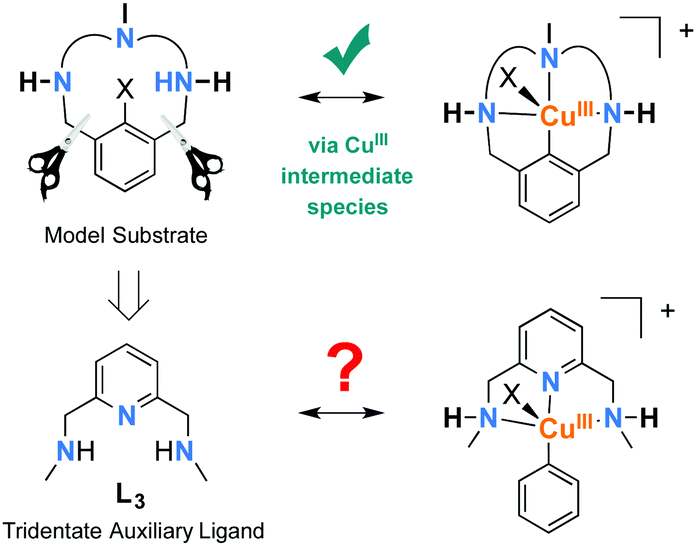
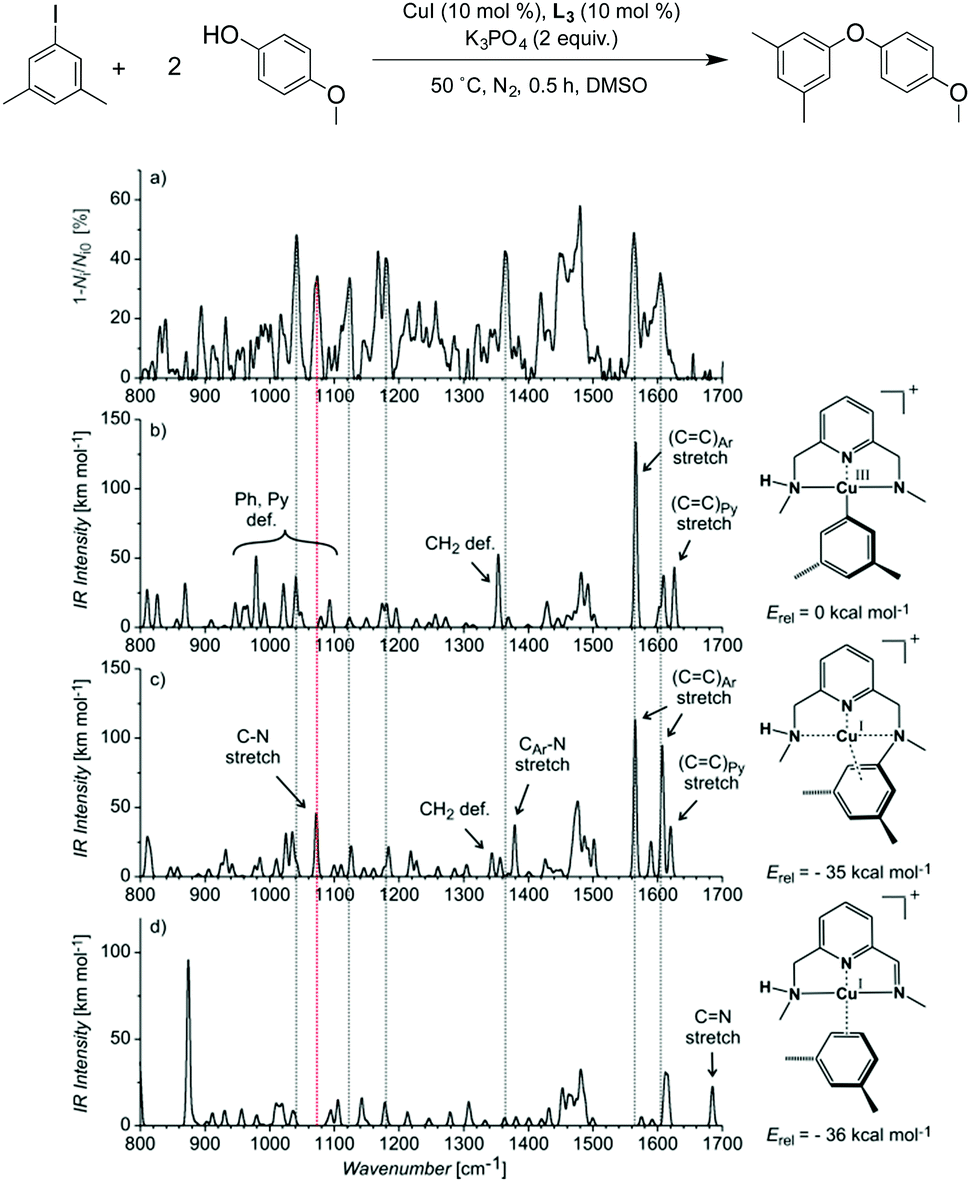
In an effort to validate whether this peak was a possible intermediate in the reaction mechanism, we extensively investigated the crude reaction mixture after 0.5 hour by characterizing the mass-selected ions at m/z = 332.1167 formed when using L3 and 1-iodo-3,5-dimethylbenzene (Fig. S4 in the ESI†) by helium-tagging infrared photodissociation (IRPD) spectroscopy.20–22 IRPD spectroscopy provides well-resolved infrared spectra of mass-selected ions.23 The experimental IRPD spectrum of ions with m/z 332 and the theoretical spectra of possible complexes corresponding to the same mass are shown in Fig. 2. We assumed that the detected ions could correspond to the proposed ([L3′–CuIII–(C6H3Me2)]+) intermediate. The theoretical spectrum of this copper(III) complex, however, does not contain any band that could be attributed to the experimental peak at 1070 cm−1 (highlighted by the red line in Fig. 2). The search for alternative structures resulted in finding a more stable species with the same mass, in which the phenyl group migrated to the nitrogen atom forming a complex of copper(I) with the N-phenylated ligand [(L3–C6H3Me2)–CuI]+ (Fig. 2c). The peculiar band at 1070 cm−1 can be assigned to the N–C (aromatic) stretching band of this complex. This result was further confirmed by measuring the IRPD spectrum of an authentic sample of [(L3–C6H5)–CuI]+ (complexation of the independently synthesized ligand L3–C6H5 and CuI). This spectrum is identical to the IRPD spectrum of the ions found in the reaction mixture for the Ullmann coupling of iodobenzene with p-methoxyphenol using the copper catalyst with L3 (Fig. 3).
 | ||
| Fig. 2 Experimental He-IRPD spectrum of ions with m/z 332 isolated from the crude reaction mixture after 0.5 h (a), compared to the theoretical IR spectra (B3LYP-D3/6-31G**(Cu:6-311G*), scaling factor: 0.98) of other plausible species involved in the reaction (b–d). | ||
 | ||
| Fig. 3 Experimental He-IRPD spectrum of (a) the isolated peak m/z 304 from the crude reaction mixture after 0.5 h, compared to the experimental IRPD spectrum of (b) an authentic sample of [(L3–C6H5)–CuI]+. | ||
When [(L3–C6H5)–CuI]+ was identified in the reaction mixture, we immediately considered the possibility that it could be the actual catalyst of C–O coupling. However, this was discarded based on our experimental findings. The use of the independently synthesized [(L3–C6H5)–CuI]+ complex as a catalyst (10 mol%) for the arylation of 2 afforded 16% yield of the biaryl ether product, compared to 45% yield when using [L3–CuI]+ under the same catalytic conditions (Fig. S20, ESI†). A similar drop in yield was found for the arylation of benzamide (from 75% down to 29%) and cyclohexylamine (from 21% down to 0%).
In light of these results, we propose that the CuI/CuIII cycle may be the main operative mechanism of this coupling reaction, the detected species being the intramolecular reductive elimination product ([(L3–C6H5)–CuI]+) of the putative [L3′–CuIII–(C6H5)]+ species, i.e. a decomposition product of the active catalyst. This non-radical mechanism may involve the formation of the [L3–CuI]+ active species followed by a low-barrier aryl iodide oxidative addition and deprotonation of one of the secondary amines to produce [L3′–CuIII–(aryl)]I. At this reaction crossroad, an axial ligand exchange by the nucleophile may trigger reductive elimination to afford the diaryl ether coupling product, or destabilization of the CuIII complex can induce intramolecular reductive elimination, yielding the CuI amine-arylation by-product detected by MS.
DFT calculations were performed to unravel the mechanism of formation of the [(L3–C6H5)–CuI]+ complex (Fig. 4a). The grouping of the aryl iodide adduct (B), which is coordinated in η2 fashion to the copper center d(Cu–Cipso) = 1.999 Å and d(Cu–Cortho) = 2.045 Å, is endergonic (ΔG = 5.8 kcal mol−1). However, the B adduct easily undergoes oxidative addition to the [L3–CuIII–(C6H5)]+ species (D) through the transition state C, which is only 1.6 kcal mol−1 higher in energy than B (i.e. ΔG# = 7.4 kcal mol−1). Removal of an iodine anion and NH deprotonation of D affords the [L3′–CuIII–(C6H5)]+ intermediate (E). Finally, E proceeds to the detected species [(L3–C6H5)–CuI]+ (G) through the reductive elimination transition state F with a barrier of 10.5 kcal mol−1. Transition state F involves a conformational change of the aryl ligand to achieve a proper geometrical orientation that favours the aryl–N coupling. Precedents of arylation of amines are known using well-defined macrocyclic aryl–CuIII model systems5 and standard Ullmann systems.19 DFT calculations predict that the geometry adopted by the copper centre significantly varies depending on the oxidation state of the metal (Fig. S18 and S19, ESI†). Further experimental substantiation of this structural variability is provided by the crystal structures of the tetrahedral [(L3)CuI(Br)]n and square-pyramidal (tetragonally distorted) [(L3)CuII(OTf)2](OTf) complexes shown in Fig. S18 and S19 (ESI†).
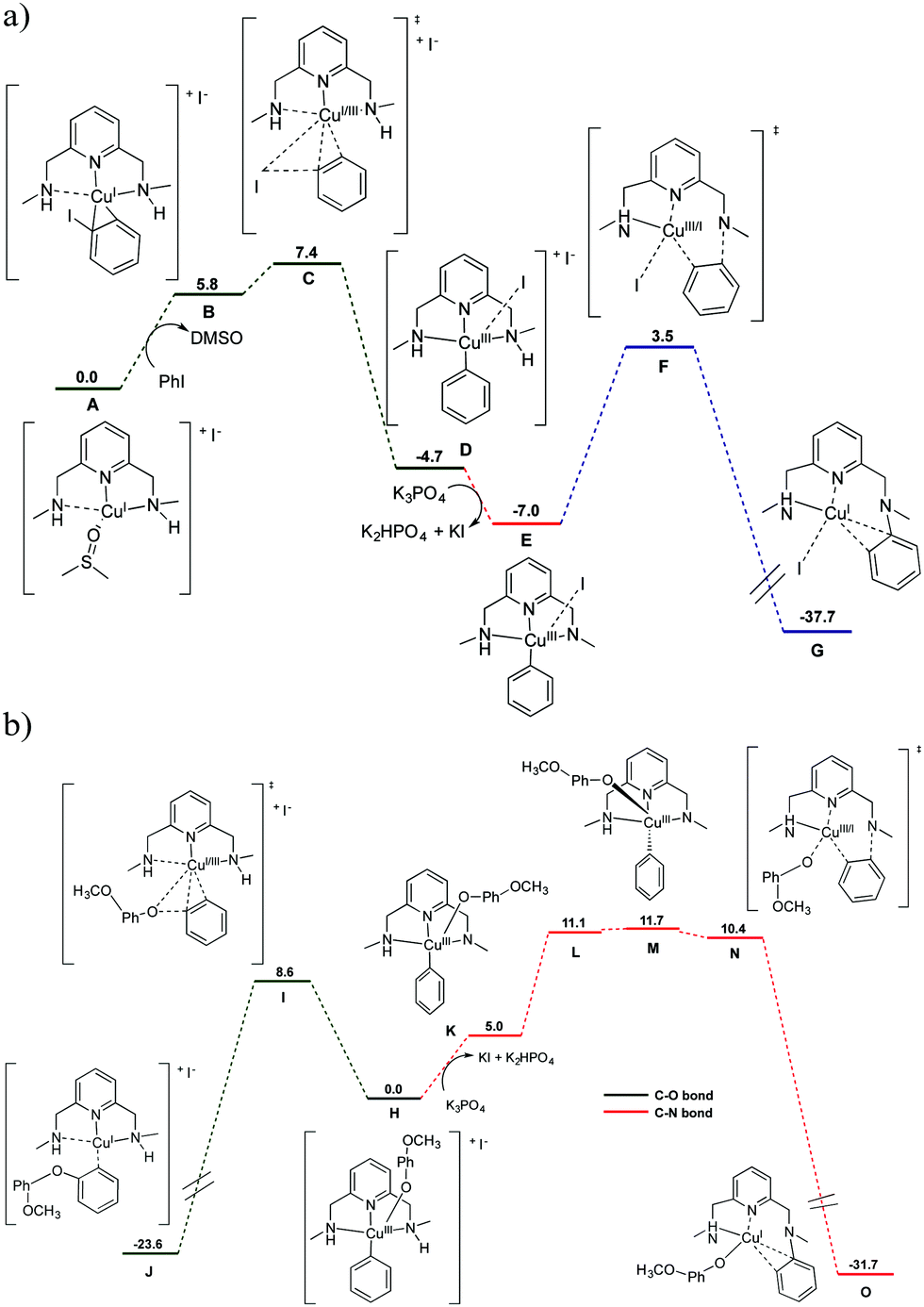 | ||
| Fig. 4 (a) DFT (B3LYP/cc-pVTZ,I:SDD+d+f) free energy profile (ΔG in kcal mol−1) for the reaction of [L3–CuI(DMSO)]I (species A) with PhI in the presence of K3PO4 as an external base in DMSO solvent (SMD model). The K3PO4, K2HPO4, KI, PhI and DMSO molecules are included in the calculation to ensure the correct energetic balance in all the reaction steps. (b) Free energy profile for the competing aryl–O coupling towards product formation and aryl–N coupling towards catalyst deactivation. See ESI† for full description of the computational details. | ||
In the presence of p-methoxyphenolate, theoretical calculations predict a favourable aryl–O coupling to form the biaryl ether product (with a free energy barrier of 8.6 kcal mol−1). The competing aryl–N coupling leading to catalyst decomposition proceeds via a higher energy barrier (11.7 kcal mol−1; Fig. 4b). The presence of phenolate makes the aryl–N coupling less favourable, because it turns nitrogen deprotonation, i.e. the step preceding C–N coupling, from an exergonic to an endergonic process (−2.3 kcal mol−1 for the D → E transformation in the absence of phenolate vs. 5.0 kcal mol−1 for the H → K transformation when phenolate is present; compare Fig. 4a and b). After amine deprotonation, K first undergoes the required conformational change of the aryl ligand that has a free energy cost of 6.7 kcal mol−1 (M). Finally, the very reactive intermediate M proceeds to the detected species O through a barrierless reductive elimination transition state (N). Mechanistic insights on the selectivity between C–O and C–N couplings in Ullmann catalysis have been documented.24–26
Therefore, a CuI/CuIII mechanism underlying the coupling reaction of iodobenzene and phenol derivatives stems from the above experimental and theoretical data; further proof was obtained by conducting the coupling experiment using the radical clock 1-allyloxy-2-iodobenzene (rc) as the substrate and p-methoxyphenol (2), benzamide (3) and cyclohexamine (4) as nucleophiles (see Scheme 2).13,27,28 The formation of the cyclized coupling compounds (rc-cyc-x) was not detected in any reaction, thus indicating the unfeasibility of a radical mechanism. We did observe relevant amounts of compound rc-H (up to 25% with cyclohexylamine 4 as a nucleophile), where the iodine atom has been substituted by an H atom. Protodecupration of a putative organometallic aryl–Cu bond has been already observed in a recent report.19
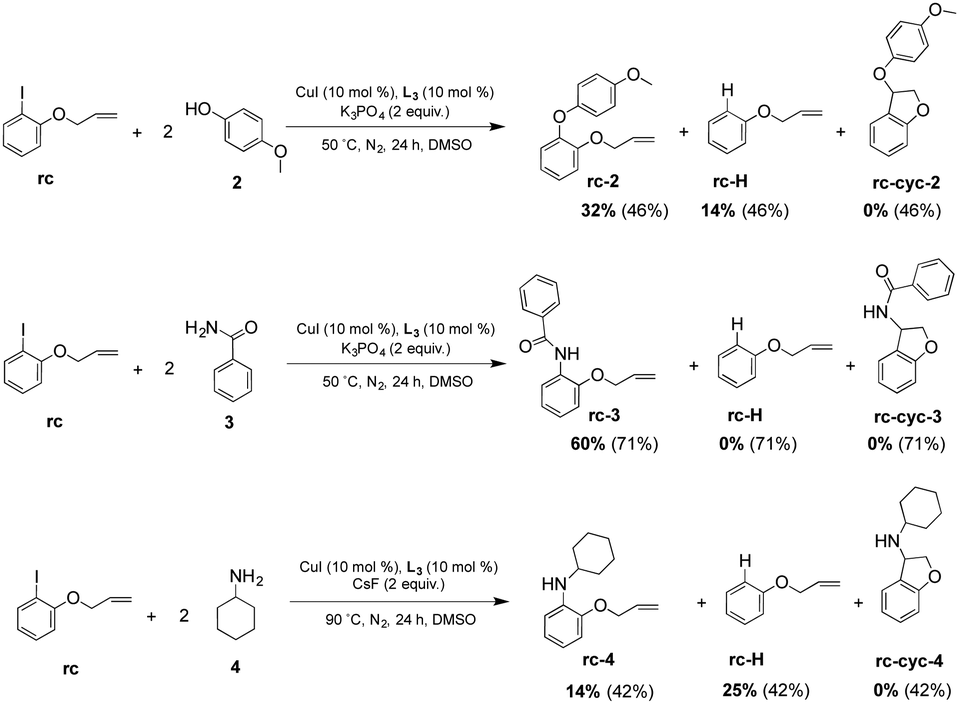 | ||
| Scheme 2 Selected coupling reactions using radical clock rc as substrate (conversion in parenthesis). | ||
In conclusion, the efficient auxiliary ligand L3 in C–O Ullmann-type couplings undergoes a decomposition pathway following a CuI/CuIII mechanism via intramolecular arylation of one of the secondary amines of the complex. The helium tagging IRPD spectroscopy and DFT mechanistic studies, along with the absence of cyclized products using an rc radical clock and the observation of protodecupration products, strongly support the existence of an aryl–CuIII species in Ullmann couplings using this specially designed tridentate L3 ligand. Insights on decomposition gained from this model system has turned out to be valuable in catalyst and process design work, shedding light on the complex chemistry of Ullmann couplings.
This work was financially supported by grants from the European Research Council (Starting Grant Project ERC-2011-StG-277801 to XR and ERC-2015-CoG-682275 (IsoMS) to JR), the Spanish MICINN (CTQ2016-77989-P to XR and CTQ2014-52525-P to JML), and the Catalan DIUE of the Generalitat de Catalunya (2014SGR862). We thank Prof. J. Lloret-Fillol (ICIQ-Tarragona) for fruitful discussions. X. R. thanks ICREA for an ICREA Acadèmia award. We thank COST Action CHAOS (CA15106) and STR from UdG for technical support.
Notes and references
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC
.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed
-
A. Casitas and X. Ribas, in Copper-Mediated Cross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley & Sons, Hoboken, 2013, ch. 7, pp. 253–279 Search PubMed
- A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301–2318 RSC
-
Y. Jiang, L. Xu, C. Zhou and D. Ma, in C-H and C-X Functionalization. Transition Metal Mediation, ed. X. Ribas, RSC Publishing, Cambridge, UK, 2013, Catalysis Series, vol. 11, ch. 1, pp. 1–45 Search PubMed
- X. Jin and R. P. Davies, Catal. Sci. Technol., 2017, 7, 2110–2117 CAS
- G. O. Jones, P. Liu, K. N. Houk and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 6205–6213 CrossRef CAS PubMed
- J. W. Tye, Z. Weng, G. Giri and J. F. Hartwig, Angew. Chem., Int. Ed., 2010, 49, 2185–2189 CrossRef CAS PubMed
- R. Giri and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 15860–15863 CrossRef CAS PubMed
- J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971–9983 CrossRef CAS PubMed
- S.-L. Zhang, L. Liu, Y. Fu and Q.-X. Guo, Organometallics, 2007, 26, 4546–4554 CrossRef CAS
- S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647–651 CrossRef CAS PubMed
- Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed
- H.-Q. Do, S. Bachman, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 2162–2167 CrossRef CAS PubMed
- A. Casitas, M. Canta, M. Solà, M. Costas and X. Ribas, J. Am. Chem. Soc., 2011, 133, 19386–19392 CrossRef CAS PubMed
- A. Casitas, A. E. King, T. Parella, M. Costas, S. S. Stahl and X. Ribas, Chem. Sci., 2010, 1, 326–330 RSC
- K. Yamaguchi, J. Mass Spectrom., 2003, 38, 473–490 CrossRef CAS PubMed
- M. Rovira, M. Soler, I. Güell, M.-Z. Wang, L. Gómez and X. Ribas, J. Org. Chem., 2016, 81, 7315–7325 CrossRef CAS PubMed
- J. Jašík, D. Gerlich and J. Roithová, J. Phys. Chem. A, 2015, 119, 2532–2542 CrossRef PubMed
- J. Jašík, J. Žabka, J. Roithová and D. Gerlich, Int. J. Mass Spectrom., 2013, 354–355, 204–210 CrossRef
- J. Roithová, Chem. Soc. Rev., 2012, 41, 547–559 RSC
- J. Jašík, D. Gerlich and J. Roithová, J. Am. Chem. Soc., 2014, 136, 2960–2962 CrossRef PubMed
- H.-Z. Yu, Y.-Y. Jiang, Y. Fu and L. Liu, J. Am. Chem. Soc., 2010, 132, 18078–18091 CrossRef CAS PubMed
- G. Lefèvre, G. Franc, A. Tlili, C. Adamo, M. Taillefer, I. Ciofini and A. Jutand, Organometallics, 2012, 31, 7694–7707 CrossRef
- A. Ouali, M. Taillefer, J.-F. Spindler and A. Jutand, Organometallics, 2007, 26, 65–74 CrossRef CAS
- E. Sperotto, G. P. M. V. Klink, G. V. Koten and J. G. D. Vries, Dalton Trans., 2010, 39, 10338–10351 RSC
- W. R. Bowman, H. Heaney and P. H. G. Smith, Tetrahedron Lett., 1984, 25, 5821–5824 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: IRPD spectroscopic studies, DFT calculations and crystallographic information. CCDC 1054559 (L3·2HCl), 1054555 ([(L3)CuI(Br)]n) and 1054556 ([L3–CuII(OTf)2]). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc04491g |
| This journal is © The Royal Society of Chemistry 2017 |