
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic role of vacancy diffusion in ceria supported atomic gold catalyst†
Zhong-Kang
Han
 ab,
Yang-Gang
Wang
*c and
Yi
Gao
*ad
ab,
Yang-Gang
Wang
*c and
Yi
Gao
*ad
aDivision of Interfacial Water and Key Laboratory of Interfacial Physics and Technology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201800 China. E-mail: gaoyi@sinap.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049 China
cFritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin-Dahlem, 14195 Germany. E-mail: wangygtccl@gmail.com
dShanghai Science Research Center, Chinese Academy of Sciences, Shanghai, 201204 China
First published on 17th July 2017
Abstract
Dynamics of intrinsic defects are considered fundamental in the chemistry of reducible oxides, and their effect on catalytic reactions have been rarely reported. Herein, we propose a new Ov diffusion assisted Langmuir–Hinshelwood mechanism for CO oxidation, which may largely account for the origin of high reactivity of supported atomic gold catalysts.
The prominent redox properties have enabled the use of reducible oxides, such as titania (TiO2) and ceria (CeO2), as important support materials or catalysts in heterogeneous catalysis.1–6 The intrinsic defects such as oxygen vacancies (Ovs), interstitials and surface steps, which ubiquitously exist in reducible oxides, have been extensively examined in literature.7–15 However, few studies are devoted to discussing their effect on the realistic catalytic reactions. This is primarily because of the complexity of the systems with multiple local minima with respect to the sites on which the excess electrons-driving the Ce4+ (4f0) to Ce3+ (4f1) reduction-localize.8,9,16 In this study, we chose ceria supported Au adatoms (monomer and dimer) as typical examples and carried out thorough density-functional theory investigations to explore the dynamics of intrinsic defects accompanying the redistribution of the 4f localized electronic states, and illustrated their key role in catalysing CO oxidation processes.
Ceria-supported gold catalysis has attracted extensive interest in the past decade due to its excellent performance in three-way catalysis, low temperature water–gas shift reaction, dehydrogenation reaction, and other catalytic applications.17–20 The high reactivity is generally attributed to the strong metal support interaction between ceria and gold particles.21,22 In particular, Ovs, the most common defects in ceria, can engender excess charges to the system, which not only enhance the binding of gold particles, but also adjust the oxidation state of gold particles.5,23 However, controversies still exist concerning the role of Ov in gold catalysis. First, on one hand, most studies reported that gold cluster preferentially occupied the surface Ov of the support.4,24–27 On the other hand, recent studies showed that the subsurface Ov was more stable than the surface Ov and was the dominant species on reduced ceria surface.28,29 Second, extensive studies have shown that the presence of oxygen vacancy in ceria support can significantly increase the activity of gold catalysis.4,30–33 However, Weststrate et al.27 recently showed that highly dispersed gold adatoms on reduced ceria seem to bind molecular CO more weakly than oxidized ceria. Camellone et al.5 demonstrated that the oxygen vacancy provides an inhibiting effect on the activity of supported gold catalysts but this effect weakens as the cluster size of gold increases. Therefore, an in-depth understanding of the interplay between gold adatoms and near-surface Ovs is of fundamental importance to explore the nature of high reactivity of supported gold catalysts.
The calculations have been performed within the framework of DFT with generalized gradient approximation using the VASP code.34,35 The DFT+U methodology was used in this study, which has been extensively utilized for ceria in literature.9,16,23,36–38 Previous studies indicated that multiple self-consistent solutions exist, corresponding to different occupations of the m projections associated with the f-shell to which the U parameter is applied.39,40 However, in this study, the used Davidson-block iteration scheme with random initialization of the orbitals, for which the most extensive tests have been performed, is considerably robust41 (see calculation details in ESI†).
To validate our methodology, the types and distributions of Ovs in the bare ceria slab (Fig. S1, S2 and Tables S1, S2, ESI†) were further examined. The most stable configurations for single surface Ov (SSV) and single subsurface Ov (SSSV) on CeO2(111) surface contain two excess electrons both located at the next-nearest neighbour cerium positions relative to the vacancy site itself. The calculated Ov formation energy of SSSV (1.91 eV) is 0.19 eV lower than that of SSV (2.10 eV), suggesting that SSSV is energetically more stable than SSV (Table S1, ESI†). This is quantitatively consistent with previous calculations (0.18 eV).9 In addition, our further examinations indicate that the vacancies tend to separate up to a distance equal to twice of the (1 × 1) surface lattice parameter (Fig. S1 and Table S1, ESI†), i.e., the third-nearest neighbour in the subsurface layer, to form oxygen vacancy pairs, which is consistent with the recent experiments and calculations.28,29
Prior to investigating the reaction mechanism of CO oxidation, we consider the interplay between gold adatoms and Ovs (Fig. S3–S5 and Tables S3, S4, ESI†), which is generally believed to be important to promote the catalytic activity of gold. For a gold monomer on CeO2(111) support with one Ov (SSV or SSSV), the most stable configurations are shown in Fig. S4a and c, ESI.† It is found that the gold monomer occupied SSV is much more stable than the SSSV with gold monomer binding to the surface oxygen, though SSSV is more stable without the presence of the gold monomer. Specifically, we find that the SSSV only needs to experience a small energy barrier of 0.27 eV to hop up from subsurface to surface site in the presence of a single gold adatom. During the hopping process, the number of excess electrons at Ce sites is reduced from 3 to 1, suggesting that the charge state of gold adatom changes from +1 to −1 by attaining two excess electrons. These results are consistent with previous calculations.4,5,12,23,42,43
We further introduce another Au atom and an extra Ov into the system to explore the effect of high concentration of vacancies on the behaviour of gold cluster. The most stable configurations for Au2@CeO2(111)-SS-SS, Au2@CeO2(111)-SS-S and Au2@CeO2(111)-S-S (SS means subsurface Ov and S means surface Ov) are listed in Fig. 1. We find an interesting reconstruction process of Au dimer in the presence of Ov pair. Initially, Au dimer is put on the surface bridged oxygen ions and the two Ov are located in the subsurface layer. The system only has 4 excess electrons corresponding to the two Ov and the charge state of Au dimer is roughly estimated to be neutral. It is shown that one of the two Ov could easily hop up from the subsurface to the surface and be filled immediately by one Au atom. This process only needs an extremely small energy barrier of 0.04 eV (TS1 in Fig. 1). Simultaneously, it will reduce the oxidation state of the gold atom, which fills into the surface Ov, from 0 to −1 but the other Au is oxidized from 0 to +1. Further hopping of the other Ov with a barrier of 0.88 eV (see TS2 in Fig. 1) is relatively difficult, partially due to the strong Coulombic repulsion of two Au-ions when both the Ov locate on the surface. These results strongly imply that under realistic conditions, not all the excess charges from Ovs can have an impact on the gold particles due to the thermodynamically unfavorable Coulombic repulsion. Compared to the gold monomer, the presence of Au dimer cluster seems to improve the stability of subsurface Ov. The energy difference for three distributions of Ov shown in Fig. 1 is less than 0.2 eV.
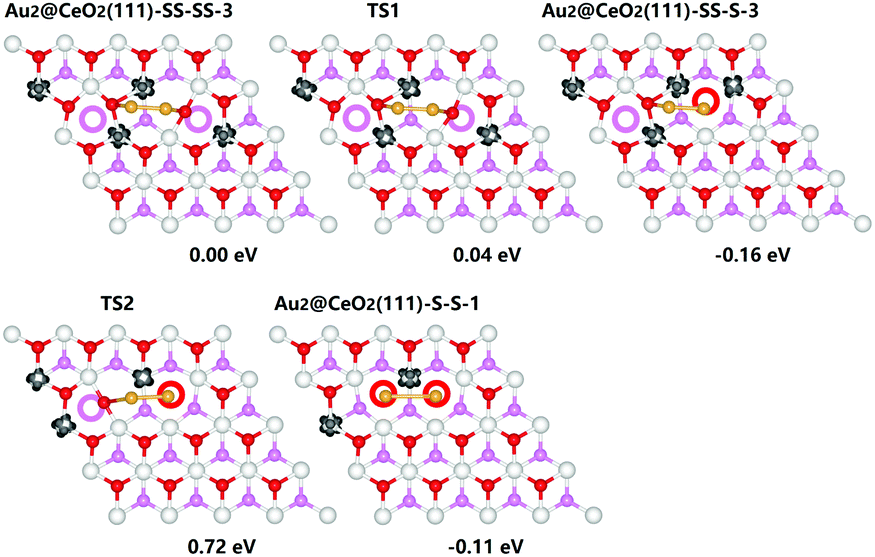 | ||
| Fig. 1 Calculated path and corresponding energetics for the migration of Ov on Au2@CeO2(111)-SS(S)-SS(S)-N. In CeO2(111)-SS(S)-SS(S)-N, SS(S) means subsurface (surface) Ov and N means they are in the nth nearest neighbour position. The spin density isosurface is shown in dark area. Cerium, surface oxygen, and subsurface oxygen atoms are depicted in white, red, and pink, respectively. Open circles represent oxygen vacancies. For clarity, only the first O−Ce−O trilayer (TL) is shown. | ||
Based on the most stable configurations of ceria-supported Au adatoms, we now consider the reactivity of CO oxidation to explore the potential effect of oxygen vacancy-metal interplay on the catalytic reactivity of reducible oxide supported metal particles. For Au monomer in Ov, the adsorption of CO is very weak (Ead = −0.16 eV) due to the full-occupied 6s orbitals of negatively charged Au, consistent with previous studies.5 Therefore, next, we only consider the reaction mechanisms of CO oxidation on ceria-supported gold dimer, where one Ov locates at the subsurface layer and the other locates at the surface layer. We find a novel Langmuir–Hinshelwood (LH) mechanism assisted by the hopping of oxygen vacancy between the surface layer and subsurface layer, which exhibits significantly higher reactivity than the traditional LH mechanism. Note that the Mars–van Krevelen Mechanism was also tested for existence but did not prove to be prevalent in this study (Fig. S6, ESI†).
In the traditional L–H mechanism (see Fig. 2a), reaction only involves CO reacting with the adsorbed O2 species and the Ovs are not allowed to diffuse during the entire process. Initially, CO is adsorbed on the gold dimer with adsorption energy of −1.58 eV but O2 molecule only weakly binds to one Au atom. The adsorption energy is −0.31 eV and the bond length of adsorbed O2 is 1.24 Å, close to 1.23 Å in the gas phase, suggesting a physisorption of O2. Next, CO and O2 move towards each other, forming an O–C–O–O* complex. This step experiences a barrier of 0.68 eV and is slightly endothermic by 0.12 eV. Subsequent formation of a gas-phase CO2 is highly exothermic (−2.17 eV). The residual O* can directly react with an additional gas-phase CO to form CO2 molecule, completing the reaction cycle.
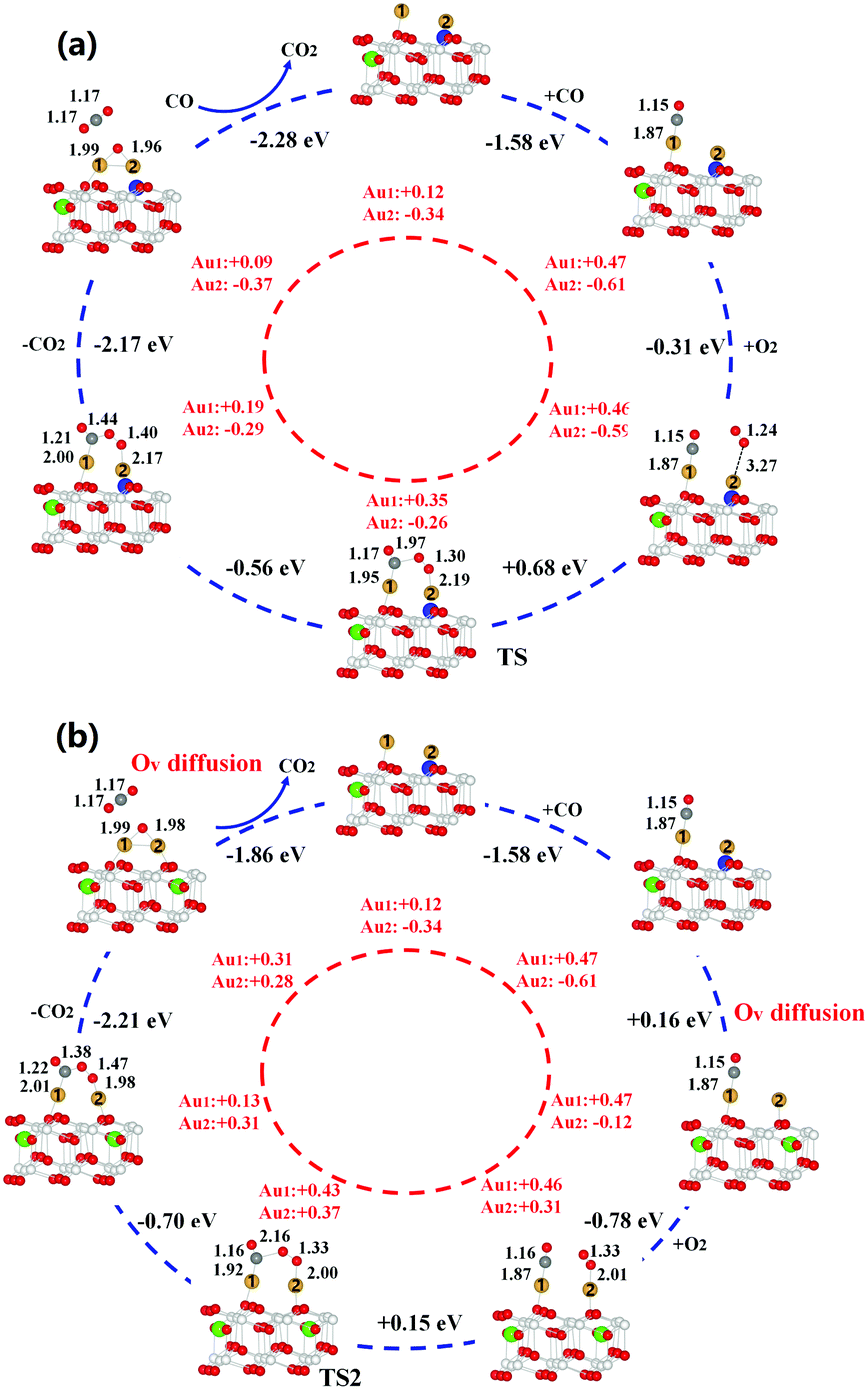 | ||
| Fig. 2 Catalytic cycle and corresponding charge cycle for the oxidation of CO catalysed by Au2@CeO2(111) via the traditional (a) and novel (b) L–H reaction mechanisms. The charges corresponding to gold adatoms are labeled as 1 and 2, respectively. Colour schemes: Au, gold; Ce, white; surface O, red; subsurface O, pink; surface Ov, blue; subsurface Ov, green; C, grey. | ||
In contrast, Fig. 2b shows a dynamic reaction mechanism of CO oxidation assisted by the oxygen vacancy diffusion. CO initially adsorbs to the Au dimer with high adsorption energy of −1.58 eV. Distinctly different from the traditional L–H mechanism, O2 adsorption is significantly enhanced by 0.31 eV, when the surface Ov diffuses into the subsurface by overcoming a small barrier of 0.29 eV. The bond length of the adsorbed O2 is increased to 1.33 Å, indicating the formation of superoxide species (O2–). After that, the adsorbed CO and O2 only need to experience a barrier of 0.15 eV to form an O–C–O–O* complex. Subsequent reactions such as CO2 formation and the removal of O by an additional CO are similar to the traditional L–H mechanism. After all the adsorbed species are removed, one of the subsurface oxygen will hop back to the surface layer, completing the catalytic cycle. Overall, Ov diffusion enhances the adsorption of O2 molecule by 0.31 eV, lowers the energy barrier of rate-determining step by 0.53 eV and stabilizes the intermediate complex by 0.98 eV. After the catalytic cycle is completed, one Ov will hop back to the surface layer, charging the Au cluster and stabilizing it. Therefore, the proposed Ov diffusion assisted L–H mechanism is expected to exhibit much higher reactivity than the traditional mechanism, which may largely contribute to the high reactivity of ceria supported gold particles at low temperatures.
To understand the nature of the enhancement effect from Ov diffusion, we first monitor the charge state of Au dimer during the catalytic cycle. It is shown that the migration of Ov from surface to subsurface leads to the re-oxidation of negatively charged gold adatom, which initially occupies the Ov site, implying that the strong interplay between ceria support and the gold adatoms may occur upon Ov diffusion. We thus further analyse the change of spin density isosurface and the projected density of states (PDOS) during the Ov diffusion processes, as shown in Fig. 3. In Fig. 3a and b, it is found that one additional 4f electron is localized on a Ce atom when the surface Ov migrates to the subsurface, indicating electron transfer from gold atom to ceria. This is further confirmed by the increase of 4f states in Fig. 3c and d. Before the diffusion, the energy levels of unoccupied O2 2π* states are higher than both saturated Au 6s states and occupied 4f states, and the O2 is thus not able to be activated. When the Ov migrates into the subsurface, the occupied Au 6s states become strongly correlated with the lattice O 2p (valence band) and as a result, the energy level of 6s gets lifted, which can release excess electrons into the Ce 4f states and unoccupied O2 2π* states. Moreover, the resultant Au 6s empty states can interact with the occupied O2 2p states, leading to a down shift of these states and stabilizing the adsorbed O2. In other words, the Ov diffusion essentially activates the Au 6s states so that they can correlate well with the frontier states of adsorbed intermediates such as O2* and O–C–O–O* complex and activate them by charge transfer.
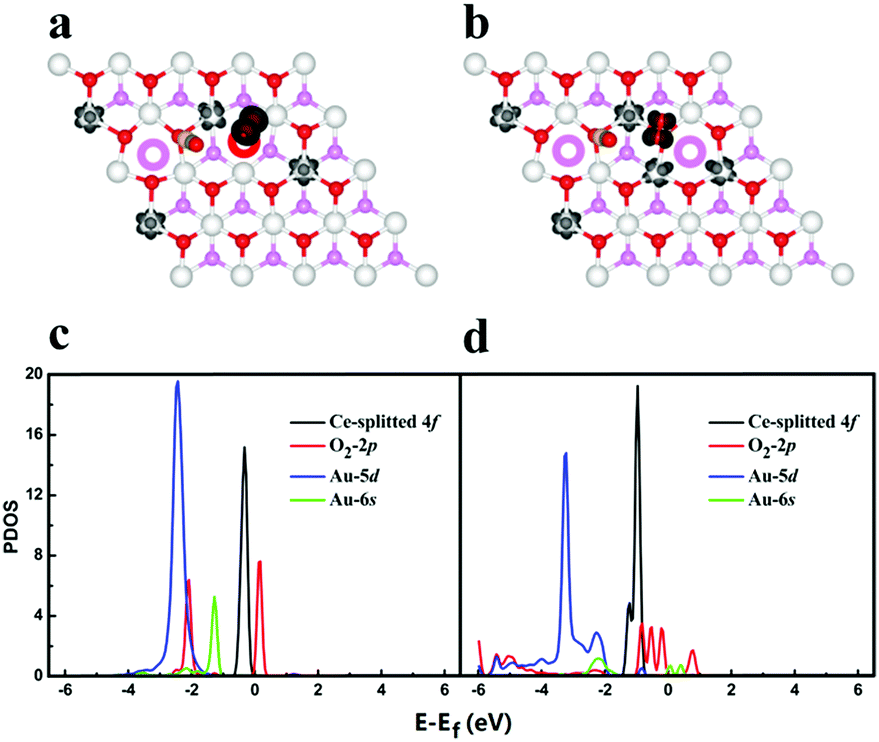 | ||
| Fig. 3 Co-adsorption of CO and O2 on Au2@CeO2(111)-SS-S-3 (a) and Au2@CeO2(111)-SS-SS-3 (b) and their projected density of state (c) and (d), respectively. The colour schemes are same with Fig. 1. | ||
In summary, we have presented a thorough understanding of charge distributions resulting from Ovs and proposed a newly dynamic L–H reaction mechanism for CO oxidation on ceria-supported Au dimer. The diffusion of Ov between the surface and subsurface layer is shown to promote the reactivity of CO oxidation during the catalytic cycle and stabilize the Au dimer after the cycle is completed. In general, this study strongly suggests that the interplay between metal particle and the intrinsic defect of the support is vital in understating the origin of high reactivity of oxide supported gold catalysis.
This study is supported by the National Natural Science Foundation of China (11574340), and the CAS-Shanghai Science Research Center (Grant No. CAS-SSRC-YJ-2015-01). Y. G. W. thanks the financial supported by Alexander von Humboldt Foundation. The authors also thank the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) under Grant No. U1501501. The computational resources utilized in this research were provided by Shanghai Supercomputer Center, National Supercomputer Centers in Tianjin, Shenzhen.
References
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- G. Deluga, J. Salge, L. Schmidt and X. Verykios, Science, 2004, 303, 993–997 CrossRef CAS PubMed.
- Z.-P. Liu, S. J. Jenkins and D. A. King, Phys. Rev. Lett., 2005, 94, 196102 CrossRef PubMed.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed.
- Y.-G. Wang, D. C. Cantu, M.-S. Lee, J. Li, V.-A. Glezakou and R. Rousseau, J. Am. Chem. Soc., 2016, 138, 10467–10476 CrossRef CAS PubMed.
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075433 CrossRef.
- M. V. Ganduglia-Pirovano, J. L. Da Silva and J. Sauer, Phys. Rev. Lett., 2009, 102, 026101 CrossRef PubMed.
- H.-Y. Li, H.-F. Wang, X.-Q. Gong, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- Y.-G. Wang, D. Mei, J. Li and R. Rousseau, J. Phys. Chem. C, 2013, 117, 23082–23089 CAS.
- Y.-G. Wang, Y. Yoon, V.-A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS PubMed.
- Z.-K. Han and Y. Gao, Nanoscale, 2015, 7, 308–316 RSC.
- M. Moser, V. Paunovic, Z. Guo, L. Szentmiklosi, M. G. Hevia, M. Higham, N. Lopez, D. Teschner and J. Perez-Ramirez, Chem. Sci., 2016, 7, 2996–3005 RSC.
- D. H. Barrett, M. S. Scurrell, C. B. Rodella, B. Diaz, D. G. Billing and P. J. Franklyn, Chem. Sci., 2016, 7, 6815–6823 RSC.
- Z. K. Han and Y. Gao, Chem. – Eur. J., 2016, 22, 2092–2099 CrossRef CAS PubMed.
- H.-Y. Li, H.-F. Wang, Y.-L. Guo, G.-Z. Lu and P. Hu, Chem. Commun., 2011, 47, 6105–6107 RSC.
- D. Andreeva, R. Nedyalkova, L. Ilieva and M. Abrashev, Appl. Catal., A, 2003, 246, 29–38 CrossRef CAS.
- C. Zhang, A. Michaelides and S. J. Jenkins, Phys. Chem. Chem. Phys., 2011, 13, 22–33 RSC.
- M. Wang, F. Wang, J. Ma, M. Li, Z. Zhang, Y. Wang, X. Zhang and J. Xu, Chem. Commun., 2014, 50, 292–294 RSC.
- X. J. Jin, K. Taniguchi, K. Yamaguchi and N. Mizuno, Chem. Sci., 2016, 7, 5371–5383 RSC.
- D. Widmann and R. J. Behm, Angew. Chem., Int. Ed., 2011, 50, 10241–10245 CrossRef CAS PubMed.
- D. Widmann and R. J. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS PubMed.
- C. Zhang, A. Michaelides, D. A. King and S. J. Jenkins, J. Am. Chem. Soc., 2010, 132, 2175–2182 CrossRef CAS PubMed.
- T. Akita, M. Okumura, K. Tanaka, M. Kohyama and M. Haruta, J. Mater. Sci., 2005, 40, 3101–3106 CrossRef CAS.
- J. A. Rodriguez, P. Liu, J. Hrbek, J. Evans and M. Perez, Angew. Chem., Int. Ed., 2007, 119, 1351–1354 CrossRef.
- J.-L. Lu, H.-J. Gao, S. Shaikhutdinov and H.-J. Freund, Catal. Lett., 2007, 114, 8–16 CrossRef CAS.
- C. Weststrate, R. Westerstrom, E. Lundgren, A. Mikkelsen, J. N. Andersen and A. Resta, J. Phys. Chem. C, 2008, 113, 724–728 Search PubMed.
- S. Torbrügge, M. Reichling, A. Ishiyama, S. Morita and O. Custance, Phys. Rev. Lett., 2007, 99, 056101 CrossRef PubMed.
- G. E. Murgida and M. V. Ganduglia-Pirovano, Phys. Rev. Lett., 2013, 110, 246101 CrossRef PubMed.
- T. Tabakova, F. Boccuzzi, M. Manzoli and D. Andreeva, Appl. Catal., A, 2003, 252, 385–397 CrossRef CAS.
- J. Rodriguez, M. Perez, J. Evans, G. Liu and J. Hrbek, J. Chem. Phys., 2005, 122, 241101 CrossRef CAS PubMed.
- A. Lindblad, R. Fink, H. Bergersen, M. Lundwall, T. Rander, R. Feifel, G. Öhrwall, M. Tchaplyguine, U. Hergenhahn and S. Svensson, J. Chem. Phys., 2005, 123, 211101 CrossRef CAS PubMed.
- R. Burch, Phys. Chem. Chem. Phys., 2006, 8, 5483–5500 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- M. Nolan, J. E. Fearon and G. W. Watson, Solid State Ionics, 2006, 177, 3069–3074 CrossRef CAS.
- B. Meredig, A. Thompson, H. Hansen, C. Wolverton and A. Van, de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195128 CrossRef.
- J. Rabone and M. Krack, Comput. Mater. Sci., 2013, 71, 157–164 CrossRef CAS.
- G. E. Murgida, V. Ferrari, M. V. Ganduglia-Pirovano and A. M. Lois, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 115120 CrossRef.
- Y. Pan, N. Nilius, H.-J. Freund, J. Paier, C. Penschke and J. Sauer, Phys. Rev. Lett., 2013, 111, 206101 CrossRef PubMed.
- M. a. M. Branda, N. C. Hernández, J. F. Sanz and F. Illas, J. Phys. Chem. C, 2010, 114, 1934–1941 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Multiple localization of the excesses 4f electrons. See DOI: 10.1039/c7cc04440b |
| This journal is © The Royal Society of Chemistry 2017 |