
Radiofluorination of a NHC–PF5 adduct: toward new probes for 18F PET imaging†
Boris
Vabre‡
a,
Kantapat
Chansaenpak‡
bc,
Mengzhe
Wang
b,
Hui
Wang
b,
Zibo
Li§
 *b and
François P.
Gabbaï§
*a
*b and
François P.
Gabbaï§
*a
aDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: gabbai@mail.chem.tamu.edu
bDepartment of Radiology, Biomedical Research Imaging Center, University of North Carolina, Chapel Hill 27599, USA. E-mail: ziboli@med.unc.edu
cNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), 111 Thailand Science Park, Pathum Thani, 12120, Thailand
First published on 11th July 2017
Abstract
The radiofluorination of N-heterocyclic carbene (NHC) phosphorus(V) fluoride adducts has been investigated. The results show that the IMe-PF5 derivative (IMe = 1,3-dimethylimidazol-2-ylidene) undergoes a Lewis acid promoted 18F–19F isotopic exchange. The resulting radiofluorinated probe is remarkably resistant to hydrolysis both in vitro as well as in vivo.
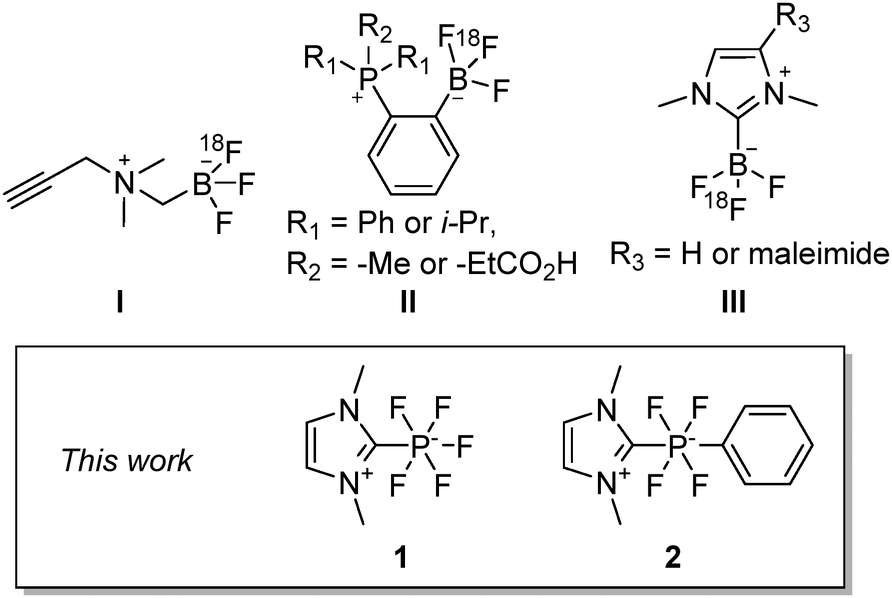
A growing area of radiochemistry is concerned with the discovery of radiolabeled prosthetic groups which, once appended to tissue- or disease-specific biomolecules, provide a modular access to novel positron emission tomography (PET) imaging agents.1 To date, most prosthetic groups contain a group 13 element2–15 or a group 14 element16–19 which serves as a binding site for the fluoride anion. Undoubtedly, boron-based prosthetic groups pioneered by Perrin are the most developed ones. The most versatile example of such a prosthetic group is the zwitterionic ammonium trifluoroborate (I) which can be incorporated in a wide range of peptide based radiotracers (Chart 1).20–25 In parallel to these advances, our interinstitutional team introduced zwitterionic phosphoniumtrifluoroborates (II)26,27 and NHC-BF3 adducts (III) which, like I, can be conjugated to biomolecules.27,28 Following up on these results, we were attracted to the fluorophilic properties of phosphorus(V) compounds.29–31 Indeed, based on computed gas phase fluoride ion affinity data (346 kJ mol−1 for BF3 and 380 kJ mol−1 for PF5), which show that P(V) fluorides31 may be more Lewis acidic than boron (III) derivatives, it occurred to us that phosphorus analogs of III might be ideally suited for application in PET.
 | ||
| Chart 1 | ||
To explore this idea and expand on the limited chemistry of radiofluorinated phosphorus compounds,32,33 we decided to investigate the radiofluorination of the N-heterocyclic carbene (NHC) phosphorus(V) fluoride derivatives 1 and 2. Compound 1 was synthesized as described in the literature. To access compound 2, we first synthesized and structurally characterized the potassium salt of the known anion [PF5Ph]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34via the “one pot” oxidation of PPhCl2 using bromine in the presence of KF (Scheme 1 and Fig. S7, ESI†). This salt, whose 19F and 31P NMR spectra are consistent with those of other [PF5Ph]− salts reported previously,34 was successfully converted into the target compound 2 in 68% yield by the addition of n-BuLi at −78 °C to a mixture of imidazolium salt and K[PF5Ph] (Scheme 1). The 19F NMR analysis of the crude mixture at room temperature after n-BuLi addition shows a doublet (JPF = 849 Hz) for 2 at −43.9 ppm and a cis product with an approximate ratio of ∼1:1 (Scheme 1). The cis adduct is characterized by three 19F resonances with a relative integration of 2:1:1, respectively. These resonances include a doublet of virtual triplets at −57.6 ppm (JPF = 783 Hz, JFF = 40 Hz) and two doublets of doublets of triplets at −43.2 ppm (JPF = 699 Hz, JFF = 49 Hz, JFF′ = 40 Hz) and −61.0 ppm (JPF = 838 Hz, JFF = 49 Hz, JFF′ = 40 Hz). Heating this mixture at 66 °C for 26 h shows isomerisation of the cis product into the trans product 2 (Fig. S5 and S6, ESI†). Compound 2 is further characterized by a 31P NMR resonance at 141.1 ppm split into a quintet (JPF = 849 Hz). The 1H NMR spectrum shows a characteristic singlet for the methyl substituents while the 13C NMR spectrum shows two doublets of quintets at 150.0 ppm (JCF = 43 Hz, JCP = 297 Hz) and 159.8 ppm (JCF = 71 Hz, JCP = 334 Hz) corresponding to the phenyl ipso-carbon and carbene carbon, respectively. These assignments align with those reported for other NHC-PF4Ph derivatives.35 The structure of 2 has been confirmed by X-ray diffraction, which shows that the carbene–phosphorus C(1)–P(1) bond (1.898(2) Å) is only slightly longer than the C(6)–P(1) bond (1.839(2)) involving the phenyl group (Fig. 1).
34via the “one pot” oxidation of PPhCl2 using bromine in the presence of KF (Scheme 1 and Fig. S7, ESI†). This salt, whose 19F and 31P NMR spectra are consistent with those of other [PF5Ph]− salts reported previously,34 was successfully converted into the target compound 2 in 68% yield by the addition of n-BuLi at −78 °C to a mixture of imidazolium salt and K[PF5Ph] (Scheme 1). The 19F NMR analysis of the crude mixture at room temperature after n-BuLi addition shows a doublet (JPF = 849 Hz) for 2 at −43.9 ppm and a cis product with an approximate ratio of ∼1:1 (Scheme 1). The cis adduct is characterized by three 19F resonances with a relative integration of 2:1:1, respectively. These resonances include a doublet of virtual triplets at −57.6 ppm (JPF = 783 Hz, JFF = 40 Hz) and two doublets of doublets of triplets at −43.2 ppm (JPF = 699 Hz, JFF = 49 Hz, JFF′ = 40 Hz) and −61.0 ppm (JPF = 838 Hz, JFF = 49 Hz, JFF′ = 40 Hz). Heating this mixture at 66 °C for 26 h shows isomerisation of the cis product into the trans product 2 (Fig. S5 and S6, ESI†). Compound 2 is further characterized by a 31P NMR resonance at 141.1 ppm split into a quintet (JPF = 849 Hz). The 1H NMR spectrum shows a characteristic singlet for the methyl substituents while the 13C NMR spectrum shows two doublets of quintets at 150.0 ppm (JCF = 43 Hz, JCP = 297 Hz) and 159.8 ppm (JCF = 71 Hz, JCP = 334 Hz) corresponding to the phenyl ipso-carbon and carbene carbon, respectively. These assignments align with those reported for other NHC-PF4Ph derivatives.35 The structure of 2 has been confirmed by X-ray diffraction, which shows that the carbene–phosphorus C(1)–P(1) bond (1.898(2) Å) is only slightly longer than the C(6)–P(1) bond (1.839(2)) involving the phenyl group (Fig. 1).
 | ||
| Scheme 1 | ||
 | ||
| Fig. 1 ORTEP diagram of 2. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): P(1)–C(6) = 1.839(2); P(1)–F(1) = 1.634(1); P(1)–F(2) = 1.642(1); P(1)–F(3) = 1.631(1); P(1)–F(4) = 1.645(1); P(1)–C(1) = 1.898(2); C(1)–P(1)–(C6) = 178.96(6); F(2)–P(1)–F(4) = 175.75(4); C(6)–P(1)–F(1) = 91.76(6). | ||
The hydrolytic stability study of 1 and 2 was evaluated using a previously published method.36 The compounds were dissolved in D2O–CD3CN (8/2 vol) at pH 7.5 ([phosphate buffer] = 500 mM) and the hydrolysis reaction was monitored by 19F NMR spectroscopy. While the salt K[PF5Ph] shows a complete hydrolysis in less than 5 min, both carbene adducts 1 and 2 are highly water stable. Compound 2 undergoes a slow hydrolysis releasing free fluoride with a pseudo-first order rate constant (kobs) of 2.3 × 10−5 min−1 (Fig. S8 and Table S3, ESI†). Surprisingly, we did not observe any free fluoride signal for 1 after five days, indicating that 1 can be considered as “eternal” (Fig. 2). It is more stable than the NHC-BF3 analogue which shows a hydrolytic rate constant (kobs) of 1.2 × 10−6 min−1 under the same conditions.28
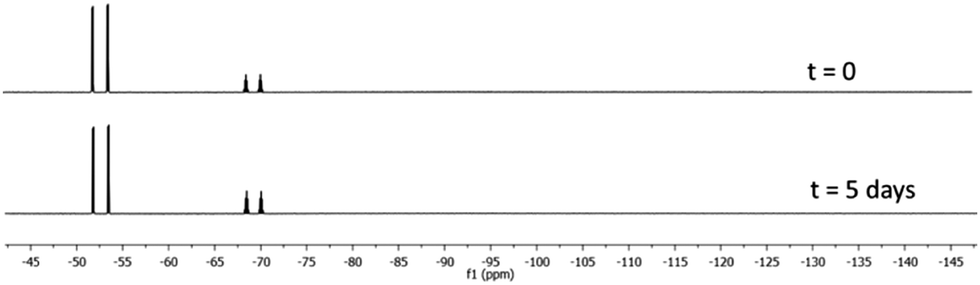 | ||
| Fig. 2 19F NMR spectrum of 1 in D2O–CD3CN (8/2 vol) phosphate buffer solution at t = 0 and t = 5 days. | ||
Next, we investigated the radiofluorination of these compounds (Scheme 2). Compound 1 could be successfully radiolabeled via18F–19F isotopic exchange using SnCl4 as a promoter, a method that we pioneered in the preparation of [18F]-BODIPY dyes.37 In this experiment, compound 1 was mixed with 5 equiv. of SnCl4 in MeCN and combined with a solution of [18F]-TBAF in the same solvent (Table 1). The reaction mixture was then shaken for 10 min at different temperatures. After being quenched by the addition of water, the radiolabeled compound ([18F]-1) was loaded on a Sep-Pak cartridge (Sep-Pak Plus tC18). Then, the excess tin reagent and by-products were removed using water. [18F]-1 was eluted off the cartridge with MeCN. A portion of the resulting MeCN solution was subjected to HPLC analysis. The identity of [18F]-1 was confirmed by the comparison of its elution time with that of its non-radioactive analog 1 (Fig. 3).
 | ||
| Scheme 2 | ||
| Entry | [1] (μmol) | SnCl4 (equiv.) | Temp. (°C) | Time (min) | SAa (mCi μmol−1) (n = 3)c | RCYb (%) (n = 3)c |
|---|---|---|---|---|---|---|
| a Specific activity is determined by dividing the product activity by the amount of the product (based on the integration of UV-HPLC and comparing with the UV chromatogram of the standard). b RCY = activity of the isolated product/starting 18F activity. All yields are decay corrected. c Each experiment was repeated 3 times. | ||||||
| 1 | 0.9 | 5 | 25 | 10 | No [18F]-1 observed | |
| 2 | 0.9 | 5 | 60 | 10 | 22.6 ± 0.6 | 4.3 ± 0.3 |
| 3 | 0.9 | 5 | 80 | 10 | 33.9 ± 1.5 | 6.6 ± 0.4 |
 | ||
| Fig. 3 Left: UV trace of 1 as the standard reference. Right: Radio-HPLC profile of 18F-[1]. | ||
As illustrated in Table 1, the radiochemical yields (RCY) of [18F]-1, calculated based on the radio-activity of the isolated product and the starting radio-activity, are quite low (4–6% decay corrected RCY). These low yields originate from the stability of the P–F bonds which impedes the 18F–19F isotopic exchange process. We found that increasing the reaction temperature leads to higher radiochemical yields (entries 1–3). However, when a high reaction temperature (100 °C) was employed, [18F]-1 was not detected by either of the two HPLC modalities (radio and UV), indicating precursor decomposition (Fig. S9). Similar issues were encountered in the radiofluorination of 2, for which all efforts proved unsuccessful including those involving different types of activators such as SnCl2, SnCl4, TMSOTf, HCl, and KHF2.
The stability of [18F]-1 was first investigated in phosphate buffer solution (0.01 M, pH 7). [18F]-1 displayed >98% radiochemical purity even after an incubation time of 3 hours. This result suggested that [18F]-1 might be extremely stable under physiological conditions. The stability of [18F]-1 was further evaluated in a murine model. The probe [18F]-1 (0.1 mCi) was injected into female nude mice and static microPET scans were obtained at 3 hours after the injection. As shown in Fig. 4, the microPET/CT images showed an obvious localization in the bladder indicating that [18F]-1 was cleared through the urinary track. More importantly, no bone uptake was observed suggesting that the [18F]-fluoride release was insignificant even 3 hours post-injection.
 | ||
| Fig. 4 Decay-corrected whole-body microPET-CT images of nude mice from a static scan at 3 h after injection of [18F]-1. (A) Coronal image and (B) sagittal image. | ||
In conclusion, we report an organophosphorus [18F]-radiotracer based on a N-heterocyclic carbene. Owing to Coulombic effects between the imidazolium and phosphate moieties, this probe is remarkably resistant to hydrolysis. It can nevertheless be radiolabeled by isotopic exchange when SnCl4 is used as an acidic promoter and can be imaged using PET for as long as three hours post-injection. We are now exploring ways to functionalize this adduct such that it can be used as a prosthetic group for targeted tissue and disease imaging.
This work was supported by the Cancer Prevention Research Institute of Texas (RP130604), Texas A&M University (Arthur E. Martell Chair of Chemistry), the National Cancer Institute (P30-CA016086-35-37), and the Biomedical Research Imaging Center, University of North Carolina at Chapel Hill.
Notes and references
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- D. M. Perrin, Acc. Chem. Res., 2016, 49, 1333–1343 CrossRef CAS PubMed.
- K. Chansaenpak, B. Vabre and F. P. Gabbai, Chem. Soc. Rev., 2016, 45, 954–971 RSC.
- Z. B. Liu, Y. Li, J. Lozada, P. Schaffer, M. J. Adam, T. J. Ruth and D. M. Perrin, Angew. Chem., Int. Ed., 2013, 52, 2303–2307 CrossRef CAS PubMed.
- Z. Liu, N. Hundal-Jabal, M. Wong, D. Yapp, K. S. Lin, F. Benard and D. M. Perrin, MedChemComm, 2014, 5, 171–179 RSC.
- Y. Li, R. Ting, C. W. Harwig, d. K. U. auf, C. L. Bellac, P. F. Lange, J. A. H. Inkster, P. Schaffer, M. J. Adam, T. J. Ruth, C. M. Overall and D. M. Perrin, MedChemComm, 2011, 2, 942–949 RSC.
- U. A. D. Keller, C. L. Bellac, Y. Li, Y. M. Lou, P. F. Lange, R. Ting, C. Harwig, R. Kappelhoff, S. Dedhar, M. J. Adam, T. J. Ruth, F. Benard, D. M. Perrin and C. M. Overall, Cancer Res., 2010, 70, 7562–7569 CrossRef PubMed.
- R. Ting, C. Harwig, U. auf dem Keller, S. McCormick, P. Austin, C. M. Overall, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2008, 130, 12045–12055 CrossRef CAS PubMed.
- R. Ting, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2005, 127, 13094–13095 CrossRef CAS PubMed.
- W. J. McBride, D. M. Goldenberg and R. M. Sharkey, J. Nucl. Med., 2014, 55, 1043 CrossRef PubMed.
- W. J. McBride, C. A. D'Souza, R. M. Sharkey and D. M. Goldenberg, Appl. Radiat. Isot., 2012, 70, 200–204 CrossRef CAS PubMed.
- W. J. McBride, R. M. Sharkey, H. Karacay, C. A. D'Souza, E. A. Rossi, P. Laverman, C.-H. Chang, O. C. Boerman and D. M. Goldenberg, J. Nucl. Med., 2009, 50, 991–998 CrossRef CAS PubMed.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Sanderson and G. Reid, Chem. – Eur. J., 2015, 21, 4688–4694 CrossRef CAS PubMed.
- R. Bhalla, C. Darby, W. Levason, S. K. Luthra, G. McRobbie, G. Reid, G. Sanderson and W. Zhang, Chem. Sci., 2014, 5, 381–391 RSC.
- A. Khoshnevisan, M. Jauregui-Osoro, K. Shaw, J. B. Torres, J. D. Young, N. K. Ramakrishnan, A. Jackson, G. E. Smith, A. D. Gee and P. J. Blower, EJNMMI Res., 2016, 6, 34 CrossRef PubMed.
- V. Bernard-Gauthier, J. J. Bailey, Z. Liu, B. Wängler, C. Wängler, K. Jurkschat, D. M. Perrin and R. Schirrmacher, Bioconjugate Chem., 2016, 27, 267–279 CrossRef CAS PubMed.
- V. Bernard-Gauthier, J. J. Bailey, Z. Liu, B. Wängler, C. Wängler, K. Jurkschat, D. M. Perrin and R. Schirrmacher, Bioconjugate Chem., 2016, 27, 267–279 CrossRef CAS PubMed.
- E. Schirrmacher, B. Wängler, M. Cypryk, G. Bradtmöller, M. Schäfer, M. Eisenhut, K. Jurkschat and R. Schirrmacher, Bioconjugate Chem., 2007, 18, 2085–2089 CrossRef CAS PubMed.
- R. Schirrmacher, G. Bradtmöller, E. Schirrmacher, O. Thews, J. Tillmanns, T. Siessmeier, H. G. Buchholz, P. Bartenstein, B. Wängler, C. M. Niemeyer and K. Jurkschat, Angew. Chem., Int. Ed., 2006, 45, 6047–6050 CrossRef CAS PubMed.
- M. Pourghiasian, Z. Liu, J. Pan, Z. Zhang, N. Colpo, K.-S. Lin, D. M. Perrin and F. Bénard, Bioorg. Med. Chem., 2015, 23, 1500–1506 CrossRef CAS PubMed.
- Z. Liu, G. Amouroux, Z. Zhang, J. Pan, N. Hundal-Jabal, N. Colpo, J. Lau, D. M. Perrin, F. Bénard and K.-S. Lin, Mol. Pharmaceutics, 2015, 12, 974–982 CrossRef CAS PubMed.
- Z. Liu, K.-S. Lin, F. Benard, M. Pourghiasian, D. O. Kiesewetter, D. M. Perrin and X. Chen, Nat. Protoc., 2015, 10, 1423–1432 CrossRef CAS PubMed.
- Z. B. Liu, M. Pourghiasian, M. A. Radtke, J. Lau, J. H. Pan, G. M. Dias, D. Yapp, K. S. Lin, F. Benard and D. M. Perrin, Angew. Chem., Int. Ed., 2014, 53, 11876–11880 CrossRef CAS PubMed.
- Z. B. Liu, M. A. Radtke, M. Q. Wong, K. S. Lin, D. T. Yapp and D. M. Perrin, Bioconjugate Chem., 2014, 25, 1951–1962 CrossRef CAS PubMed.
- Z. Liu, D. M. Perrin, M. Pourghiasian, F. Benard, J. Pan and K.-S. Lin, J. Nucl. Med., 2014, 55, 1499–1505 CrossRef CAS PubMed.
- Z. Li, K. Chansaenpak, S. Liu, C. R. Wade, H. Zhao, P. S. Conti and F. P. Gabbaï, MedChemComm, 2012, 3, 1305–1308 RSC.
- K. Chansaenpak, M. Wang, S. Liu, Z. Wu, H. Yuan, P. S. Conti, Z. Li and F. P. Gabbai, RSC Adv., 2016, 6, 23126–23133 RSC.
- K. Chansaenpak, M. Wang, Z. Wu, R. Zaman, Z. Li and F. P. Gabbai, Chem. Commun., 2015, 51, 12439–12442 RSC.
- T. W. Hudnall, Y.-M. Kim, M. W. P. Bebbington, D. Bourissou and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890–10891 CrossRef CAS PubMed.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- A. R. Studenov, M. J. Adam, J. S. Wilson and T. J. Ruth, J. Labelled Compd. Radiopharm., 2005, 48, 497–500 CrossRef CAS.
- M. F. Ghorab and J. M. Winfield, J. Fluorine Chem., 1990, 49, 367–383 CrossRef CAS.
- R. Schmutzler, J. Am. Chem. Soc., 1964, 86, 4500–4502 CrossRef CAS.
- T. Böttcher, O. Shyshkov, M. Bremer, B. S. Bassil and G.-V. Röschenthaler, Organometallics, 2012, 31, 1278–1280 CrossRef.
- R. Ting, C. W. Harwig, J. Lo, Y. Li, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Org. Chem., 2008, 73, 4662–4670 CrossRef CAS PubMed.
- S. L. Liu, T. P. Lin, D. Li, L. Leamer, H. Shan, Z. B. Li, F. P. Gabbai and P. S. Conti, Theranostics, 2013, 3, 181–189 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, characterization and imaging data. CCDC 1504579 and 1504580. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc04402j |
| ‡ Contributed equally to the work. |
| § Jointly conceived the study. |
| This journal is © The Royal Society of Chemistry 2017 |