
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA naphthalene-fused dimer of an anti-aromatic expanded isophlorin†
Baddigam Kiran
Reddy‡
 a,
Jeff
Rawson‡
bc,
Santosh C.
Gadekar
a,
Paul
Kögerler
*bc and
Venkataramanarao G.
Anand
*a
a,
Jeff
Rawson‡
bc,
Santosh C.
Gadekar
a,
Paul
Kögerler
*bc and
Venkataramanarao G.
Anand
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Pune, 411008, Maharashtra, India. E-mail: vg.anand@iiserpune.ac.in
bPGI-6, Forschungszentrum Jülich, 52428 Jülich, Germany
cInstitute of Inorganic Chemistry, RWTH Aachen University, 52074 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de
First published on 23rd June 2017
Abstract
We report the first synthesis of a covalent expanded isophlorin dimer from two 24-π doubly S-confused sapphyrin-like pentathiaisophlorins. It exhibits marginal peripheral aromaticity rather than strong global diatropicity or paratropicity and weak intermacrocycle electronic communication. Quantum chemical methods discern that cross-conjugation is responsible for these unusual electronic features.
Aromatic macrocycles such as porphyrin and corrole have attracted significant attention for their unique electronic properties.1 The oxidative coupling of these individual macrocyclic units, through their meso and β carbons, is a frequently employed synthetic strategy to blend them into short oligomers.2 Multiple intermolecular connectivities enhance the electronic coupling between these adjacent π sub-units leading to exceptional properties such as globally delocalized2b and electronically excited states,3 hole/electron-polaron states,4 low energy two-photon absorptions5 and low resistances when sandwiched between metal layers.6 Compared to the versatile chemistry of aromatic building blocks, inadequate synthetic protocols for structurally similar anti-aromatic macrocycles have hindered the fusion of individual 4nπ molecules into π-conjugated materials.7 Even though stable anti-aromatic isophlorinoids hold promise to explore the electronic properties of unheralded fused 4nπ systems, the enigmatic incompatibility of a meso-free isophlorin to oxidative coupling has precluded the direct synthesis of a meso–meso linked isophlorin dimer.8 Herein, we report the synthesis of the first fused dimer of an anti-aromatic expanded isophlorin along with its structural and electronic properties.
A recent report established reversible two-electron oxidation of a stable anti-aromatic isophlorin.9 Prior to this report, a stable expanded isophlorin, 1, with 25π electrons was identified as a neutral and stable radical under ambient conditions.10 It undergoes a facile one-electron oxidation and reduction to generate 24π and 26π diamagnetic species, respectively. Similar, but smaller, organic π radicals exhibit dimerization either through covalent or non-covalent interactions (Scheme 1).
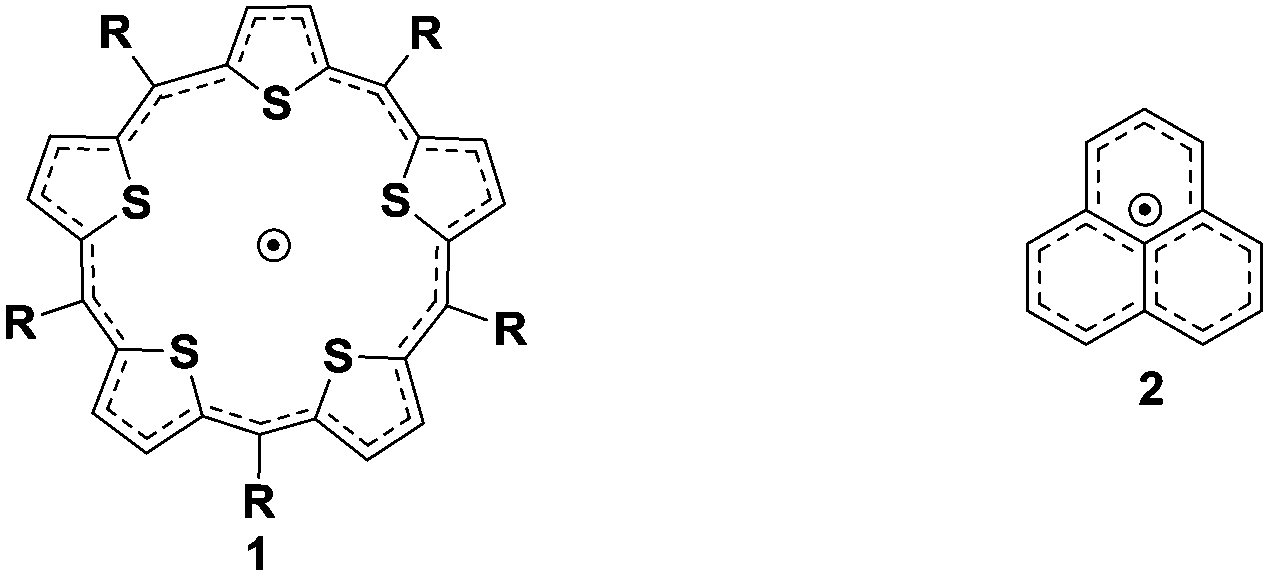 | ||
| Scheme 1 Pentathiophene 25π expanded isophlorin (1) and spin delocalization in the phenalenyl radical (2). | ||
NMR spectroscopic studies of a phenalenyl radical, 2, confirmed a non-covalent diamagnetic π-dimer at low temperatures.11 Its sigma dimer also exhibits diamagnetism when fused directly in the absence of any spacer. However, the covalent dimer can exist both as a diamagnetic and paramagnetic species depending on the nature of the link between the two phenalenyl units.12 Dimerizing 1 through one of its meso carbons offers the possibility of a biradical or a closed-shell double bond bridge between the individual macrocyclic units. Further amalgamation through the β positions should yield a completely fused expanded isophlorin dimer. Our recent report on the synthesis of a meso–meso linked 20π isophlorin dimer established that a bridging double bond results in the formation of unstable biradical species.8 Such structure-dependent spin states for dimers of expanded porphyrinoids or isophlorinoids have not been explored to date. Anticipating the stabilization of a triple-link between the two 25π pentathiophene radicals, 1, we designed the synthesis of 3 with appropriate precursors from thiophene units (Scheme 2).
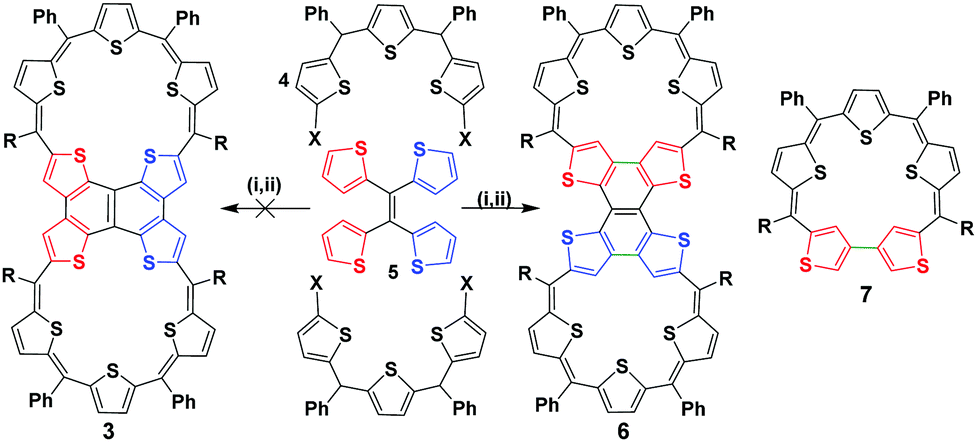 | ||
| Scheme 2 Acid-catalysed synthesis of expanded isophlorin 6. Reaction conditions: (i) BF3·OEt2 (1 eq.), dry DCM (100 ml), rt, N2, 2 h (ii) FeCl3 (10 eq.), 2 h. X = –CH(OH)R, R = C6F5. 7 represents the structural isomer of a doubly confused pentathiasapphyrin. | ||
The synthesis involved the condensation of dicarbinol, 4, with tetrathienylethene, 5,13 under acidic conditions followed by oxidation to obtain the desired expanded isophlorin dimer. A pink band was isolated from the reaction mixture through silica gel column chromatography with dichloromethane and hexane as the eluent. After evaporation of the solvent under vacuum, the obtained green solid (in 10% yield) displayed an m/z value of 1911.7206 corresponding to the mass of a triply linked dimer 3 from MALDI-TOF-TOF mass analysis. This confirmed both cyclization and the expected oxidative dehydrogenation of the thiophene beta carbons in the presence of FeCl3. Owing to the limited solubility of the isolated dimer 6, attempts to crystallize it in various solvent systems went futile until we were successful by adding a few drops of TFA in a solution of chloroform. The molecular structure as determined from single crystal X-ray diffraction analysis revealed a novel and an unusual structural isomer of the expected dimer. This revealed the unexpected condensation of the vicinal thiophene units, instead of the geminal thiophenes, with two units of diol to form the macrocyclic dimer (Fig. 1).
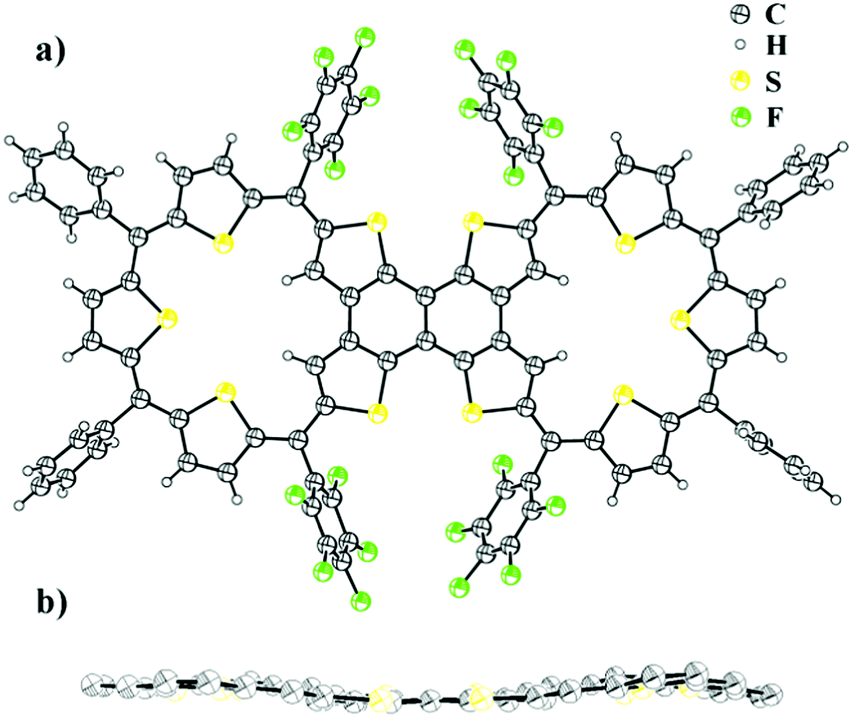 | ||
| Fig. 1 (a) Molecular structure of 6 determined from single crystal X-ray diffraction. (b) Side view of compound 6 (meso-substituents omitted for clarity). | ||
A rare β–β oxidative dehydrogenation between the adjacent two ring-inverted thiophene rings yields a sapphyrin-like framework to complete the macrocyclic framework. This unexpected fusion resembles a typical [3+2] condensation employed in the synthesis of ring-inverted sapphyrin.14
Surprisingly, the desired dimer, 3, of a 25π radical was not identified in this reaction. In spite of a large structure, with ten thiophene units, the system sustains a relatively flat topology. Compound 6 displayed a near planar structure and the crystal packing shows weak π–π interaction with an inter-planar distance of 3.79 Å between adjacent layers (see ESI†). The meso phenyl rings have a near orthogonal orientation with respect to the mean plane of the dimer. The ethylene moiety can be a part of aromatic conjugation with only one of the two macrocyclic units. Therefore, it can be considered to be a merger of an aromatic (26π electrons) and an anti-aromatic unit (24π electrons). Yet, the total number of 50π electrons corresponds to Hückel's (4n + 2) π-electrons and hence expected to display a diatropic ring current effect. The photocyclization of tetrathienylethene 5 with 366 nm light in the presence of iodine has been shown to give tetrathienonaphthalene,13 which constitutes a substructure within the framework of 6, which has limited solubility in common organic solvents, but is modestly soluble in halobenzenes. Similar to this sub-unit, the dimer 6 has poor solubility in common organic solvents. Proton NMR spectroscopy of 6 in chlorobenzene-d5 at 400 K gives chemical shifts of the thiophene β-protons that evidence its weak macrocyclic ring current. In particular, the protons of inverted rings resonate at δ 5.78 ppm, upfield from 8.1 ppm (in bromobenzene-d5 at 403 K) for the comparable positions on tetrathienonaphthalene. The protons on the two central thiophene rings of the trithiophene sub-unit resonate as a singlet at δ 7.354 ppm, downfield from 7.09 ppm at the 3-position of thiophene. The remaining two sets of thiophene β-protons evince two doublets centered at δ 7.436 ppm and 7.289 ppm. The magnitudes of the proton chemical shifts induced by the macrocyclic ring current are substantially smaller than those observed for strongly aromatic 30π thiaporphyrinoids.7
Analysis of the DFT-calculated magnetic properties of 6 using nucleus-independent chemical shifts at a distance of 1 Å from the macrocycle plane (NICS(1))15 buttresses the proton NMR evidence of marginal aromatic ring currents in this system (Fig. 2B). While strong local magnetic shieldings are determined for points centered over the internal naphthalene (−20.35 ppm) and central thiophenes (−10.47 ppm), a value of 0.20 ppm is calculated above the centroid of each pentathiophene macrocycle. This contrasts the case of a 30π aromatic hexaphyrin, for which a strong shielding NICS(1) value is calculated at the macrocycle center (−14.85 ppm; see ESI†).
 | ||
| Fig. 2 (A) Selected regions and peak assignments from the 1H NMR spectrum of 6 determined at 400 K in chlorobenzene-d5 solvent. The star marks a sideband from a solvent peak. (B) Values of NICS(1) calculated at points along the long molecular axis. | ||
The electronic absorption spectrum of 6, collected in chlorobenzene solution, consists of a low energy ground to first singlet excited state (S0 → S1) transition spanning ∼650–850 nm with the maximum extinction coefficient ε ≈ 104, a tenfold more intense S0 → S2 peak centred at 567 nm, and a continuous manifold of ε ≈ 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 absorption from 350 to 500 nm (Fig. 3). For aromatic porphyrinoids, the S0 → S1 transition typically gives rise to a series of ε = 104 visible-range peaks (Q-band), and a single ε > 105 peak at the edge of the UV region (B-band), which may be compared with the S0 → S1 and S0 → S2 bands of 6 respectively.16 However, the strong vibronic splitting of the S0 → S2 band of 6 is very different from a typical porphyrin Soret transition, which evinces minimal vibronic satellites in spite of its great intensity.17 The electronic spectrum of 6 is suggestive of a polycyclic benzenoid hydrocarbon rather than a globally aromatic porphyrinoid. A combination of TDDFT calculations and spectral deconvolution into Gaussian peaks identifies four principle electronic transitions within the 350–850 nm wavelength range, each of which is split into three discernible vibronic sub-bands that are spaced by ∼1200 cm−1. Notably, the long-axis polarized QX transition is calculated to be nearly forbidden, and the S0 → S1 manifold is attributed to the QY transition. The Y-polarized transitions are essentially a single configuration in nature (QY, H → L: 97%; BY, H−2 → L+2: 74%), while the X-polarized transitions evince strong configuration interaction (BX, H−2 → L: 46%, H → L+2: 54%; QX, H−2 → L: 52%, H → L+2: 45%). The Q-bands of a D4h metalloporphyrin are nearly forbidden,17 but for porphyrins with extended π-electron systems18 and many conjugated porphyrin oligomers,4 symmetry breaking disrupts the configuration interaction to red shift and intensify the long axis-polarized Q-derived states. For 6, the unobservable QX band and its strong configuration interaction-pairing with BX suggest that the molecule 6 is comprised of two macrocycles that function as separate entities.
000 absorption from 350 to 500 nm (Fig. 3). For aromatic porphyrinoids, the S0 → S1 transition typically gives rise to a series of ε = 104 visible-range peaks (Q-band), and a single ε > 105 peak at the edge of the UV region (B-band), which may be compared with the S0 → S1 and S0 → S2 bands of 6 respectively.16 However, the strong vibronic splitting of the S0 → S2 band of 6 is very different from a typical porphyrin Soret transition, which evinces minimal vibronic satellites in spite of its great intensity.17 The electronic spectrum of 6 is suggestive of a polycyclic benzenoid hydrocarbon rather than a globally aromatic porphyrinoid. A combination of TDDFT calculations and spectral deconvolution into Gaussian peaks identifies four principle electronic transitions within the 350–850 nm wavelength range, each of which is split into three discernible vibronic sub-bands that are spaced by ∼1200 cm−1. Notably, the long-axis polarized QX transition is calculated to be nearly forbidden, and the S0 → S1 manifold is attributed to the QY transition. The Y-polarized transitions are essentially a single configuration in nature (QY, H → L: 97%; BY, H−2 → L+2: 74%), while the X-polarized transitions evince strong configuration interaction (BX, H−2 → L: 46%, H → L+2: 54%; QX, H−2 → L: 52%, H → L+2: 45%). The Q-bands of a D4h metalloporphyrin are nearly forbidden,17 but for porphyrins with extended π-electron systems18 and many conjugated porphyrin oligomers,4 symmetry breaking disrupts the configuration interaction to red shift and intensify the long axis-polarized Q-derived states. For 6, the unobservable QX band and its strong configuration interaction-pairing with BX suggest that the molecule 6 is comprised of two macrocycles that function as separate entities.
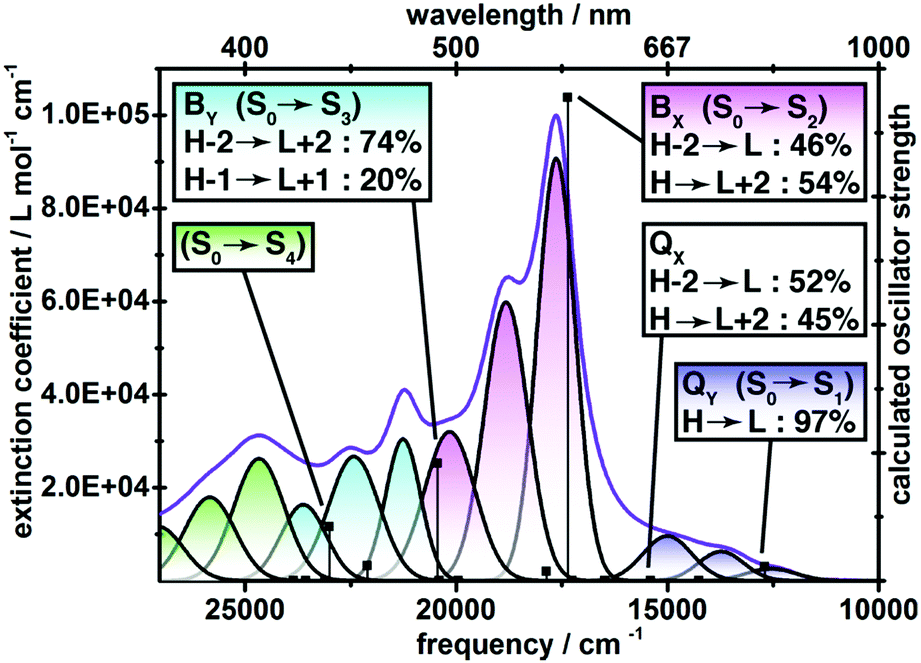 | ||
| Fig. 3 Electronic absorption spectrum of 6 measured in chlorobenzene solvent and its decomposition into Gaussian sub bands. Boxes present assignments of major transitions based upon TDDFT calculations, with phenomenological labels in parentheses, and the principle electronic configurations of each transition as determined by population analysis. | ||
The modest electronic coupling down the long axis of the molecule 6 is better understood by constructing a correlation diagram in which the bis-macrocycle is modeled as two S-confused thiasapphyrinoids 7 united by an ethylene bridge (Fig. 4). Note that the ethylene b3g and b1u wavefunctions are forbidden by symmetry from constructive overlap with the thiasapphyrinoid au and b2g wavefunctions which constitute the HOMO and LUMO of 6, respectively, and also with the progenitors of H−3 and L+3 of 6. These orbitals evince nodal planes that bisect the bridge carbons, and their energies are scarcely perturbed from those of their antecedents in the non-bonded dimer. A comparison of the TDDFT-calculated electronic transitions for 7 and 6 further shows that the spectral features evinced by 6 may be ascribed to the through-space excitonic coupling between the two thiasapphyrinoid building blocks in the weak coupling limit (see the ESI† for more details).19
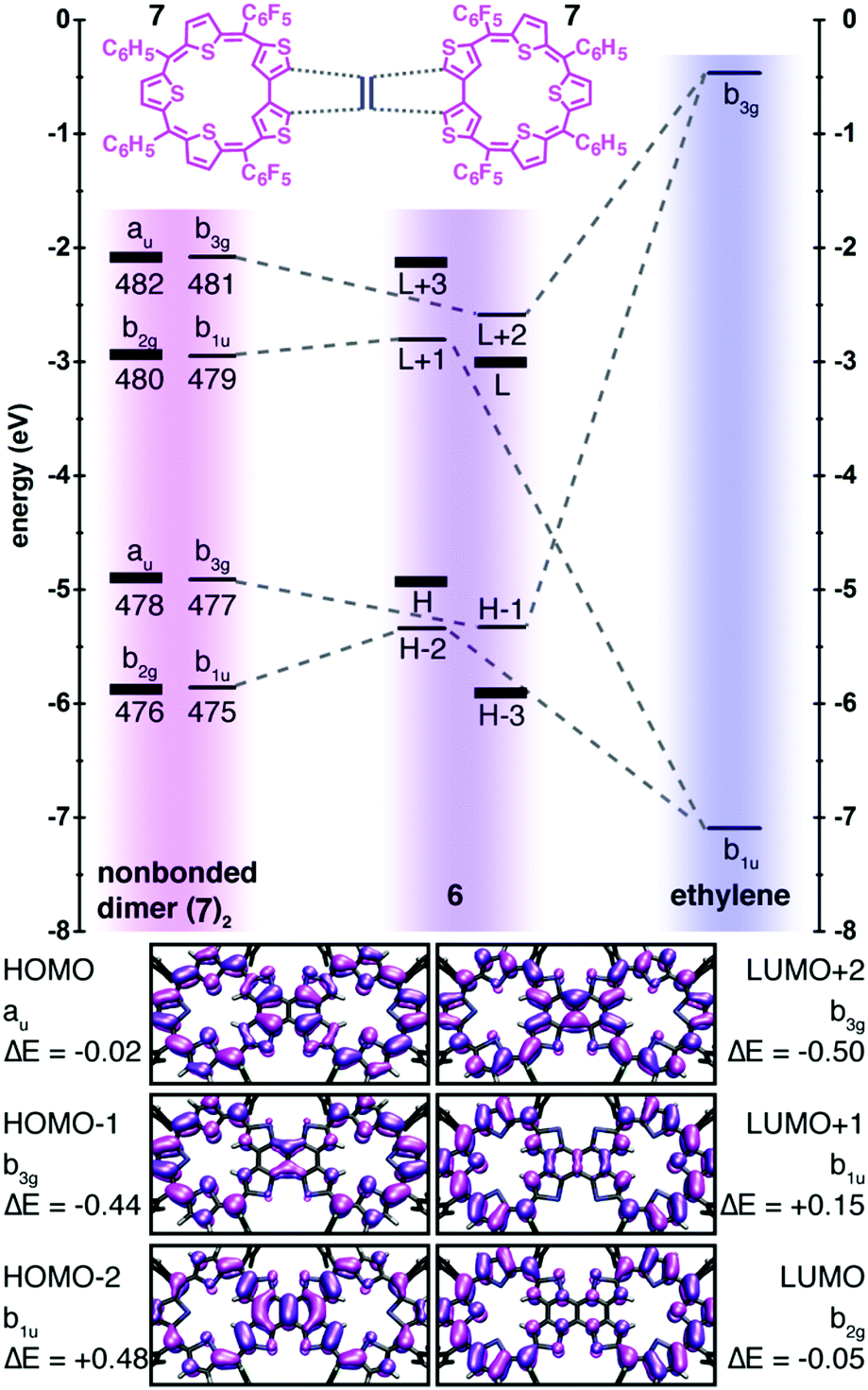 | ||
| Fig. 4 Top: Correlation diagram that deconstructs 6 into thiasapphyrinoid and ethylene fragments. Bottom: Frontier orbital wave functions of 6 plotted as 0.02 isodensity surfaces, with energy differences between them and their progenitors in the non-bonded thiasapphyrinoid dimer. | ||
The marginal global aromaticity of the macrocycles of 6 and the weak communication between them are consequences of cross-conjugation, where two unsaturated groups are conjugated to a third, but not to each other.17 The linkage of the putative thiasapphyrinoids 7 by the ethylene bridge is one such instance of cross-conjugation, as this bridge forbids any reasonable resonance structures that push electrons from one ring to the other. The structure of 7 itself contains two exocyclic double bonds at the point where thiophene rings are joined by their β-positions, constituting a doubly cross-conjugated linkage which limits its anti-aromatic character despite the 24π-electron count of its circuit. A similarly structured doubly N-confused 22π sapphyrin was indeed found to evince a negligible peripheral aromatic ring current.14 Our experimental and computational evidence supports the significance of this qualitative Lewis structure analysis, yet more desirable would be a quantitative method derived from molecular orbital calculations.
In summary, we have synthesized a novel dimer of an anti-aromatic expanded isophlorin. The synthesis for the first time reveals the oxidative coupling of inverted heterocyclic rings, in contrast to non-inverted heterocyclic rings in aromatic porphyrinoids. The impact of cross-conjugation effects upon molecular conductance resonances20 and electron transfer rates21 has been recognized recently, and this structural motif has been prominently explored in annulene chemistry.22 The decisive influence of cross-conjugation upon macrocycle aromaticity and intermolecular electronic communication outlined here provides another dramatic example of nonlinear conjugation.
Parts of this work were supported through EU ERC grant no. 308051 – MOLSPINTRON. BKR thanks UGC, New Delhi, India, for the research fellowship. VGA thanks DST, New Delhi, India, for the Swarnajayanti Fellowship.
Notes and references
- R. Guilard, J.-M. Barbe, C. Stern and K. M. Kadish, The Porphyrin Handbook, Academic Press, Amsterdam, 2003, pp. 303–349 Search PubMed.
- (a) V. Lin, S. DiMagno and M. Therien, Science, 1994, 264, 1105–1111 CAS; (b) A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed; (c) S. Ooi, T. Tanaka, K. H. Park, S. Lee, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 3107–3111 CrossRef CAS PubMed; (d) J. Sankar, H. Rath, V. Prabhuraja, S. Gokulnath, T. K. Chandrashekar, C. S. Purohit and S. Verma, Chem. – Eur. J., 2007, 13, 105–114 CrossRef CAS PubMed.
- T. V. Duncan, K. Susumu, L. E. Sinks and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 9000–9001 CrossRef CAS PubMed.
- (a) F. C. Grozema, C. Houarner-Rassin, P. Prins, L. D. A. Siebbeles and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 13370–13371 CrossRef CAS PubMed; (b) K. Susumu, P. R. Frail, P. J. Angiolillo and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 8380–8381 CrossRef CAS PubMed; (c) J. Rawson, P. J. Angiolillo and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13779–13783 CrossRef CAS PubMed.
- (a) Y. Nakamura, S. Y. Jang, T. Tanaka, N. Aratani, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Chem. – Eur. J., 2008, 14, 8279–8289 CrossRef CAS PubMed; (b) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- (a) Z. Li, T.-H. Park, J. Rawson, M. J. Therien and E. Borguet, Nano Lett., 2012, 12, 2722–2727 CrossRef CAS PubMed; (b) G. Sedghi, V. M. Garcia-Suarez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martin, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS PubMed.
- B. K. Reddy, A. Basavarajappa, M. D. Ambhore and V. G. Anand, Chem. Rev., 2017, 117, 3420–3443 CrossRef CAS PubMed.
- B. K. Reddy, S. C. Gadekar and V. G. Anand, Chem. Commun., 2016, 52, 3007–3009 RSC.
- S. P. Panchal, S. C. Gadekar and V. G. Anand, Angew. Chem., Int. Ed., 2016, 55, 7797–7800 CrossRef CAS PubMed.
- T. Y. Gopalakrishna, J. S. Reddy and V. G. Anand, Angew. Chem., Int. Ed., 2014, 53, 10984–10987 CrossRef CAS PubMed.
- K. Goto, T. Kubo, K. Yamamoto, K. Nakasuji, K. Sato, D. Shiomi, T. Takui, M. Kubota, T. Kobayashi, K. Yakusi and J. Ouyang, J. Am. Chem. Soc., 1999, 121, 1619–1620 CrossRef CAS.
- (a) S. Suzuki, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2006, 128, 2530–2531 CrossRef CAS PubMed; (b) K. Uchida, S. Ito, M. Nakano, M. Abe and T. Kubo, J. Am. Chem. Soc., 2016, 138, 2399–2410 CrossRef CAS PubMed; (c) Z. Mou, K. Uchida, T. Kubo and M. Kertesz, J. Am. Chem. Soc., 2014, 136, 18009–18022 CrossRef CAS PubMed.
- E. Fischer, J. Larsen, J. B. Christensen, M. Fourmigué, H. G. Madsen and N. Harrit, J. Org. Chem., 1996, 61, 6997–7005 CrossRef CAS PubMed.
- J. L. Sessler, D.-G. Cho, M. Stȩpień, V. Lynch, J. Waluk, Z. S. Yoon and D. Kim, J. Am. Chem. Soc., 2006, 128, 12640–12641 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- P. J. Angiolillo, J. Rawson, P. R. Frail and M. J. Therien, Chem. Commun., 2013, 49, 9722–9724 RSC.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- N. F. Phelan and M. Orchin, J. Chem. Educ., 1968, 45, 633 CrossRef CAS.
- (a) J. Lykkebo, A. Gagliardi, A. Pecchia and G. C. Solomon, ACS Nano, 2013, 7, 9183–9194 CrossRef CAS PubMed; (b) G. C. Solomon, D. Q. Andrews, R. P. Van Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 7788–7789 CrossRef CAS PubMed.
- (a) E. Maggio, G. C. Solomon and A. Troisi, ACS Nano, 2014, 8, 409–418 CrossRef CAS PubMed; (b) A. B. Ricks, G. C. Solomon, M. T. Colvin, A. M. Scott, K. Chen, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 15427–15434 CrossRef CAS PubMed.
- (a) A. Bandyopadhyay, B. Varghese, H. Hopf and S. Sankararaman, Chem. – Eur. J., 2007, 13, 3813–3821 CrossRef CAS PubMed; (b) M. Gholami and R. R. Tykwinski, Chem. Rev., 2006, 106, 4997–5027 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1476102 for X-ray crystal structure of 6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc04050d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |