
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Templated and self-limiting calcite formation directed by coccolith organic macromolecules†
Assaf
Gal
 ab,
Richard
Wirth
c,
Zahava
Barkay
d,
Noam
Eliaz
e,
André
Scheffel
a and
Damien
Faivre
*b
ab,
Richard
Wirth
c,
Zahava
Barkay
d,
Noam
Eliaz
e,
André
Scheffel
a and
Damien
Faivre
*b
aMax-Planck Institute of Molecular Plant Physiology, Potsdam-Golm 14476, Germany
bMax-Planck Institute of Colloids and Interfaces, Potsdam-Golm 14476, Germany. E-mail: Damien.Faivre@mpikg.mpg.de
cGeoForschungsZentrum Potsdam 4.3, Telegrafenberg, Potsdam 14473, Germany
dWolfson Applied Materials Research Center, Tel-Aviv University, Israel
eDepartment of Materials Science and Engineering, Tel-Aviv University, Israel
First published on 16th June 2017
Abstract
The formation of intricately shaped crystalline minerals by organisms is orchestrated by specialized biomacromolecules. The macromolecules associated with coccoliths, nanometer-sized calcite crystal arrays produced by marine microalgae, can form a distinct calcium-rich phase via macromolecular recognition. Here, we show that this calcium-rich phase can be mineralized into a thin film of single-crystalline calcite by the balanced addition of carbonate ions. Such a crystallization process provides a strategy to direct crystalline products via local interactions between soluble macromolecules and compatible templates.
The classical viewpoint of crystallization processes is centered on the interactions between solvated ions during nucleation, and between ions and crystal surfaces during growth.1,2 Such interactions are inherently stochastic, rendering the control of crystal nucleation and growth challenging. One strategy to control the nucleation of the crystalline phase is to use a template that lowers the kinetic barrier to nucleation.3–5 In many systems, both biological and synthetic, control over the location and orientation of the initial nucleus is achieved by heterogeneous nucleation from functionalized surfaces.6–10 Further crystal growth can then proceed by the attachment of ions, or even larger particles,11–13 from the surrounding solution, resulting in a crystallographic habit of the crystal. The presence of soluble macromolecules in the solution is often associated with striking disequilibrium morphologies such as thin crystalline films or tablets.10,14–16 The process underlying crystal growth into such morphologies is not well understood.
A remarkable example for biological control over crystal nucleation and growth are the calcite crystals of coccoliths. Coccoliths are mineralized organic scales that are formed inside the cells of coccolithophores, a dominant group of marine unicellular algae.17 Coccolith formation occurs inside an intracellular vesicle, where calcite crystals nucleate and grow on the rim of an organic scale called base plate.18 Soluble macromolecules are an important component inside the coccolith vesicle, and become tightly associated with the mineral phase.19 A previous study,20 investigating the coccoliths of the species Pleurochrysis carterae, showed that macromolecular recognition between the soluble macromolecules, mainly the three known polysaccharides,19 and the rim of the base plate, directs large amounts of calcium ions to the site destined for crystal formation. Interestingly, a mineral phase was not observed, raising the question of how mineralization can be achieved in the system.20 In the current study, we report the formation of calcite within this Ca-rich environment at conditions where crystallization from the solution is absent. Crystallization in these conditions is strictly restricted to the privileged microenvironment controlled by the organic macromolecules, yielding oriented thin calcite crystals on the base plate.
P. carterae coccoliths were harvested from cultured cells, and their calcite crystals dissolved. The organic fraction, base plates as well as soluble macromolecules, was retained after dialysis. The base plates are oval in shape, 1–2 μm in diameter and ∼10 nm thick, and are barely visible in a scanning electron microscope (SEM) on the supporting membrane (Fig. 1A). When calcium ions (1 mM) are added to the coccolith organic fraction, they trigger the macromolecular recognition reaction between the negatively charged soluble macromolecules and the rim of the base plates, as reported earlier.20 This reaction leads to the aggregation of calcium-loaded macromolecules at the rim of the base plate. This Ca-rich phase is not mineralized (Fig. 1B). In the present experimental procedure, aliquots of Na2CO3 solution were directly mixed into the reaction volume, following the formation of the Ca-rich phase on the coccolith base plate, with the aim of inducing mineral formation. For a wide range of carbonate concentrations the system behaved as expected for calcium carbonate precipitation in the presence of organic additives. At low concentrations of carbonate (less than 5 mM) no precipitation was observed, and at sufficiently high concentrations (more than 20 mM) uncontrolled precipitation of rhombohedral calcite occurred (Fig. 1C and Table S1, ESI†).
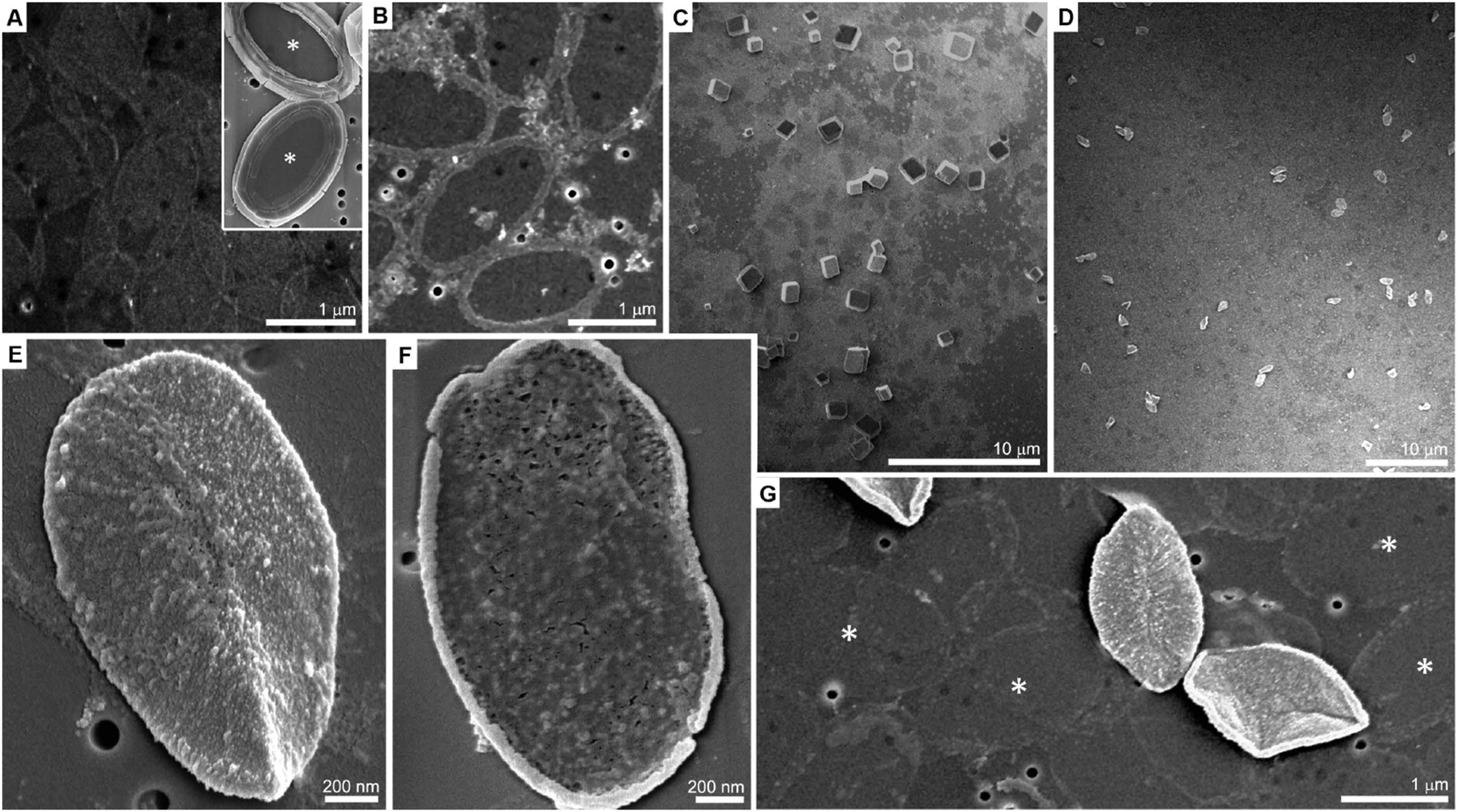 | ||
| Fig. 1 (A) SEM image of isolated base plates. The inset shows intact P. carterae coccoliths, with their baseplates indicated with asterisks. (B) When 1 mM calcium ions are added to the base plates and the soluble macromolecules, they form dense aggregates around the base plate periphery. (C) Adding 50 mM carbonate ions leads to the precipitation of rhombohedral calcite crystals. (D) However, if only 10 mM carbonate ions are added, calcite growth from solution is suppressed and only the formation of precipitates on base plates can be seen after few hours. (E and F) Higher magnification of base plates encrusted only on one side. (G) Two encrusted base plates, with opposite orientations, and many un-mineralized base plates (several are indicated with asterisks) that lost most of the Ca-loaded macromolecules at their rims. Note that for imaging with the SEM all samples were dried on a track-etched membrane. Thus, the 100 nm round holes that are visible in the images are part of the support, and not of the sample. | ||
A detailed investigation of the carbonate concentration range needed to bring the solution close to the conditions that yield precipitation revealed a concentration window in which a completely different behavior occurs (Table S1, ESI†). If carbonate is added to a concentration of 10 mM and the solution is left to ripe over the course of 4 hours, the distinctive rhombohedral calcite crystals, indicative for homogeneous nucleation and subsequent solution-mediated crystal growth, can no longer be detected. Instead, different objects of uniform size and shape are obtained (Fig. 1D). Higher-magnification micrographs show that these objects are base plates that are encrusted with a newly formed phase (Fig. 1E). A close inspection shows that only the upper, mineral-associated, side of the base plate is covered by this phase while the lower side is bare (Fig. 1F). Base plates with deposits of the new phase are a minor fraction of the total base plates present in the sample. The deposit-free base plates seem to have lost most of their initial ring composed of the un-mineralized calcium-rich phase (Fig. 1G).
We used high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) to characterize the encrusted base plates (Fig. 2A and Fig. S1, ESI†). High-magnification imaging reveals highly ordered lattice fringes that matched the crystal structure of calcite (Fig. 2B). Electron diffraction pattern from a single mineralized base plate shows a single set of diffraction spots (Fig. 2C), suggesting that the calcite is aligned. Atomic force microscope (AFM) images show that the mineral coverage of the base plate is 60–100 nm thick, with granular morphology of fused particles, 10–50 nm in diameter (Fig. 2D). The crystallographic registry of the calcified base plates was investigated using transmission Kikuchi diffraction (TKD) in SEM. Mapping the c-axis orientation throughout the mineralized base plate shows a narrow distribution around a single orientation (Fig. 2E and F). Interestingly, in all the analyzed base plates, with only one exception (9 out of 10), the c-axis orientation is ±25° deviating from the supporting film plane, and the a-axis perpendicular ±25° to it. Since the calcified base plate usually is not lying flat on the support, we attribute much of the orientation variation to the variability in sample positioning, and conclude that a common orientation of the calcite crystals relative to the base plate is observed: the c-axis lies in the base plate plane and the a-axis is vertical to it.
 | ||
| Fig. 2 (A) HAADF-STEM image of a calcified base plate. (B) HRTEM lattice fringe image showing lattice fringes with d-spacing corresponding to the calcite lattice. The inset shows the corresponding fast Fourier transform (FFT) diffraction pattern with crystallographic annotations for several spots. (C) Selected-area electron diffraction (SAED) pattern collected from an entire calcified base plate. (D) AFM height-image of a calcified base plate. (E) Color coded map of inverse pole figure of c-axis orientations of the calcite crystals in a single calcified base plate. The inset shows the SEM image of the same calcified base plate. (F) Pole figures showing the angular distribution of the a- and c-axes. Note that the c-axis is close to the stereogram circumference and, thus, almost parallel to the imaging plane, and one of the a-axes is close to the stereogram center, thus almost perpendicular to it. | ||
We investigated several aspects underlying this crystallization process, which leads to a thin single crystal of calcite. The most striking observation is that none of the mineralized base plates grew further than the ∼100 nm thin calcite cover. This cannot be growth cessation due to depletion of calcium and\or carbonate, because adding a measured amount of carbonate resulted in the mineralization of more base plates with the same morphology rather than further growth of the existing crystals (Fig. 3A and B).
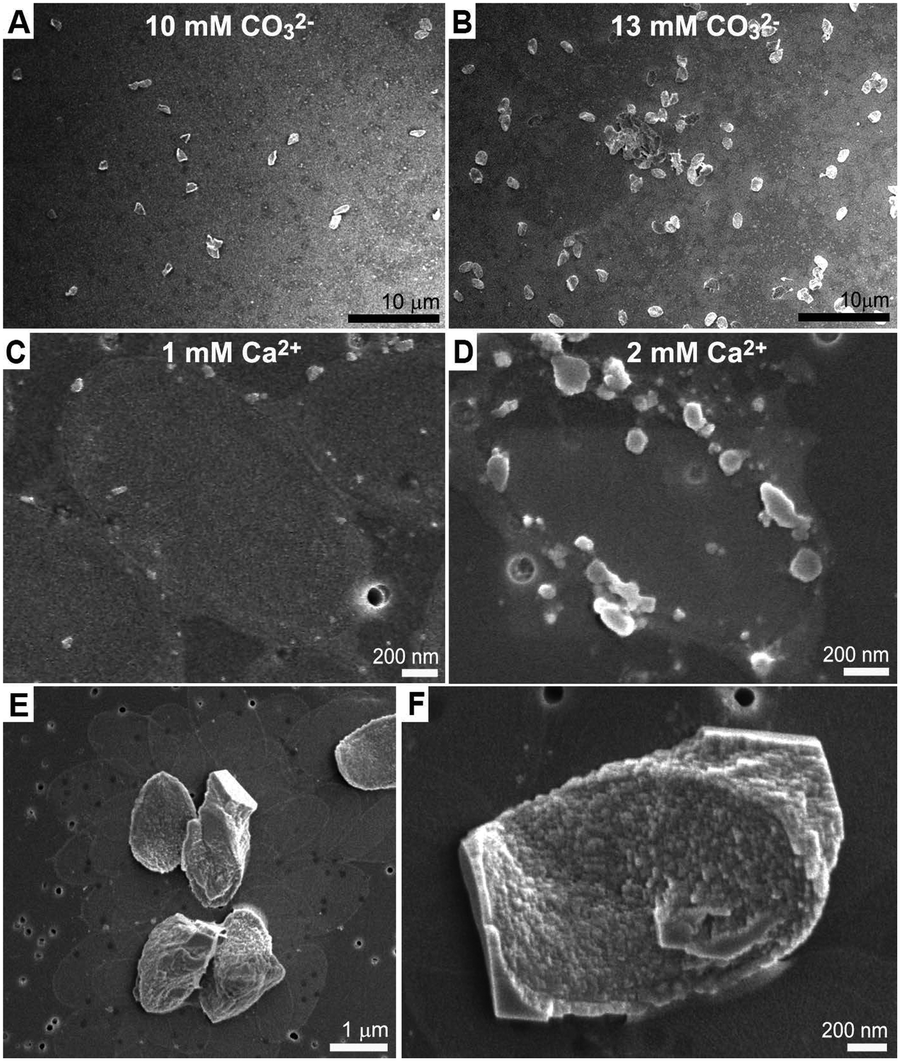 | ||
| Fig. 3 (A and B) SEM images of mineralization experiments of the same batch of coccolith organic fraction imaged after 4 hours, using two different carbonate concentrations. When more carbonate is present more base plates are mineralized, but the size of the crystals is the same. (C and D) Quenching the mineralization reaction after 10 minutes shows amorphous aggregates, more prominent at higher Ca concentrations, at the base plate rim. (E) Low- and (F) high-magnification images of crystallized base plates formed in the presence of soluble macromolecules diluted 10 times relative to the initial composition. The calcite crystals are no longer tightly covering the base plate, and develop micron-sized crystallographic facets. | ||
Quenching the reaction after 10 minutes showed that initial mineral precipitation occurs on the base plate rim in the form of an amorphous phase containing calcium and carbonate ions (Fig. 3C, D, Fig. S2 and S3, ESI†). Most likely, the crystallization proceeds from the circumference of the base plate inwards, and an amorphous precursor phase is an intermediate phase. The carbonate buffer system plays only an indirect role in this process as similar results are obtained at pH values from 8 to 11, as long as the carbonate concentration is appropriate (Table S1, ESI†). Increasing calcium concentration results in crystallization of rhombohedral crystals in addition to the base plates (Table S2, ESI†). All these observations suggest a mechanism in which crystallization is restricted only to the Ca-rich phase that is formed by the soluble macromolecules. To test this hypothesis, experiments where the soluble macromolecules were diluted with respect to the base plates were carried out. These experiments show that the restriction of the crystallization process to the base plate surface is gradually lost with the dilution, as the crystals grow outwards developing rhombohedral facets (Fig. 3E, F and Table S2, ESI†). This is in agreement with the Ca-rich phase losing its unique properties and becoming more similar to the surrounding solution.
The results of these in vitro experiments suggest that coccolith organic components can manipulate the chemical environment in their vicinity and funnel the crystallization process to a prescribed outcome via the following steps: adding calcium ions to the coccolith organic components triggers the macromolecular recognition reaction,20 and results in the formation of three distinct calcium environments (Fig. 4A and B). Some calcium ions are aggregated at the base plate periphery, another fraction is more loosely complexed by macromolecules that are still suspended in the solution, and some calcium ions are left unbound. Adding carbonate ions to this system leads to differential interactions with the different calcium environments (Fig. 4C).21 Since unbound calcium ions are abundant, if the solution saturation level is high enough, solution-mediated crystal nucleation and growth will prevail. However, we experimentally found that there is a carbonate concentration range that is not high enough to induce crystallization in the solution, yet sufficient to induce precipitation in the Ca-loaded aggregates at the base plate periphery (Fig. 4D). This crystallization process starts by heterogeneous nucleation on the base plate surface. Due to a kinetic barrier, not all the base plates nucleate crystals, and the ones that do not lose their associated calcium in a process that resembles Ostwald ripening. What is evident by the current observations is that propagation of crystal growth is restricted to the nanoscale Ca-rich phase created by the interaction of the macromolecules and calcium ions. How such dense chemical environment facilitates crystal nucleation and growth, and what chemical interactions occur between the organic and inorganic components, need to be further elucidated.
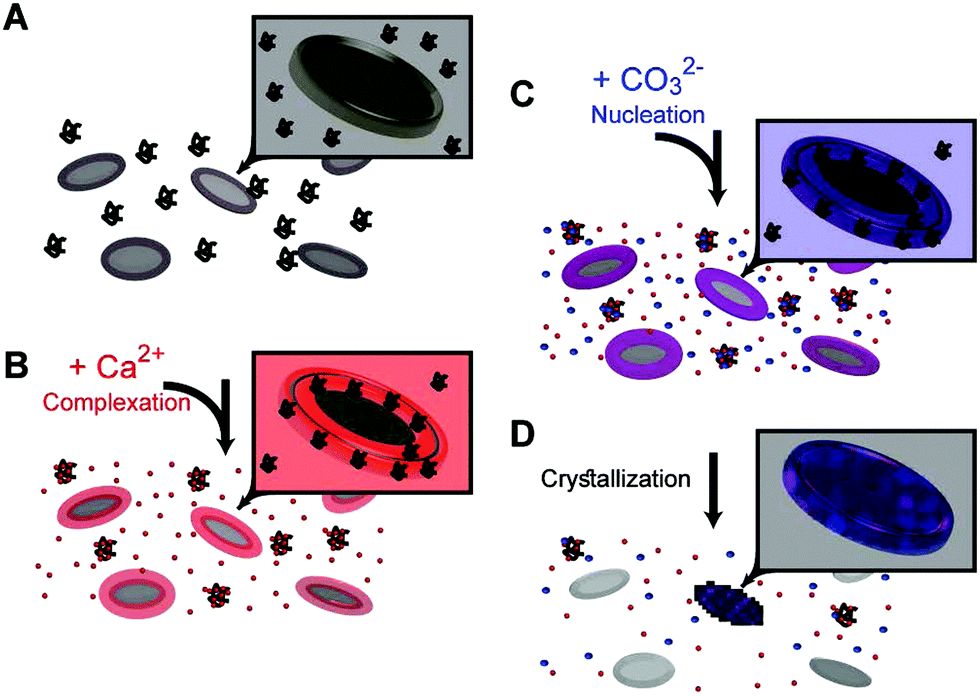 | ||
| Fig. 4 The different components and their interactions during the base plate crystallization experiments. Base plates and soluble macromolecules (represented as coils) are in black. Calcium, either as ions or dense phases, is in red. Carbonate is depicted in blue, and calcium carbonate in purple. The insets show one base plate in a detailed view. | ||
Controlling the stochastic process of crystallization in solution is highly challenging. The investigated in vitro crystallization process, using macromolecules extracted from coccoliths, points to a potentially important biological strategy. Negatively charged polymers complex calcium ions, and by means of specific attachment to a compatible surface, a microenvironment is created that is the preferred site for crystallization upon addition of the counter-ion. Such a process can inspire engineered nano-patterning of inorganic solids that is not based on the delicate balance of thermodynamic considerations of solution-mediated crystal growth, but rather on the specific interactions between polymers, recruiting the inorganic material to prescribed locations.
This research was supported by the Max-Planck Society, the Deutsche Forschungsgemeinschaft (DFG) grants Sche1637/3-1 to AS, and FA835/9-1 to DF. AG. was supported by an Alexander von Humboldt postdoctoral fellowship. Open Access funding provided by the Max Planck Society.
Notes and references
- J. W. Gibbs, Trans. Conn. Acad. Arts Sci., 1876, 2, 108–248 Search PubMed ; 343–524.
- J. J. De Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS.
- L. M. Hamm, A. J. Giuffre, N. Han, J. Tao, D. Wang, J. J. De Yoreo and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1304–1309 CrossRef CAS PubMed.
- P. Fratzl, F. D. Fischer, J. Svoboda and J. Aizenberg, Acta Biomater., 2010, 6, 1001–1005 CrossRef PubMed.
- A. J. Giuffre, L. M. Hamm, N. Han, J. J. De Yoreo and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9261–9266 CrossRef CAS PubMed.
- G. Falini, S. Albeck, S. Weiner and L. Addadi, Science, 1996, 271, 67–69 Search PubMed.
- F. Nudelman, B. A. Gotliv, L. Addadi and S. Weiner, J. Struct. Biol., 2006, 153, 176–187 CrossRef CAS PubMed.
- A. Berman, D. J. Ahn, A. Lio, M. Salmeron, A. Reichert and D. Charych, Science, 1995, 269, 515–518 CAS.
- T. Kato, A. Sugawara and N. Hosoda, Adv. Mater., 2002, 14, 869–877 CrossRef CAS.
- E. DiMasi, V. M. Patel, M. Sivakumar, M. J. Olszta, Y. P. Yang and L. B. Gower, Langmuir, 2002, 18, 8902–8909 CrossRef CAS.
- R. Q. Song and H. Colfen, Adv. Mater., 2010, 22, 1301–1330 CrossRef CAS PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Z. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Colfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- G. F. Xu, N. Yao, I. A. Aksay and J. T. Groves, J. Am. Chem. Soc., 1998, 120, 11977–11985 CrossRef CAS.
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS.
- N. Hosoda and T. Kato, Chem. Mater., 2001, 13, 688–693 CrossRef CAS.
- H. R. Thierstein and J. R. Young, Coccolithophores: from molecular processes to global impact, Springer, 2004 Search PubMed.
- J. R. Young and K. Henriksen, Rev. Mineral. Geochem., 2003, 54, 189–215 CrossRef CAS.
- M. E. Marsh, D. K. Chang and G. C. King, J. Biol. Chem., 1992, 267, 20507–20512 CAS.
- A. Gal, R. Wirth, J. Kopka, P. Fratzl, D. Faivre and A. Scheffel, Science, 2016, 353, 590–593 CrossRef CAS PubMed.
- P. J. M. Smeets, K. R. Cho, R. G. E. Kempen, N. A. J. M. Sommerdijk and J. J. De Yoreo, Nat. Mater., 2015, 14, 394–399 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional EM images and tables. See DOI: 10.1039/c7cc03639f |
| This journal is © The Royal Society of Chemistry 2017 |