
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Measurement of 14N quadrupole couplings in biomolecular solids using indirect-detection 14N solid-state NMR with DNP†
J. A.
Jarvis‡
a,
I.
Haies§
 b,
M.
Lelli¶
c,
A. J.
Rossini||
c,
I.
Kuprov
b,
M.
Carravetta
b and
P. T. F.
Williamson
*a
b,
M.
Lelli¶
c,
A. J.
Rossini||
c,
I.
Kuprov
b,
M.
Carravetta
b and
P. T. F.
Williamson
*a
aBiological Sciences, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: p.t.williamson@soton.ac.uk
bChemistry Department, University of Southampton, Southampton, SO17 1BJ, UK
cCentre de RMN à Tres Hauts Champs, Institut de Sciences Analytiques, Université de Lyon (CNRS/ENS Lyon/UCB Lyon1), 69100 Villeurbanne, France
First published on 26th October 2017
Abstract
The quadrupolar interaction experienced by the spin-1 14N nucleus is known to be extremely sensitive to local structure and dynamics. Furthermore, the 14N isotope is 99.6% naturally abundant, making it an attractive target for characterisation of nitrogen-rich biological molecules by solid-state NMR. In this study, dynamic nuclear polarization (DNP) is used in conjunction with indirect 14N detected solid-state NMR experiments to simultaneously characterise the quadrupolar interaction at multiple 14N sites in the backbone of the microcrystalline protein, GB3. Considerable variation in the quadrupolar interaction (>700 kHz) is observed throughout the protein backbone. The distribution in quadrupolar interactions observed reports on the variation in local backbone conformation and subtle differences in hydrogen-bonding; demonstrating a new route to the structural and dynamic analysis of biomolecules.
Nitrogen is an important element in chemistry due to its prevalence in biological and naturally occurring materials. However, solid-state NMR (ssNMR) studies of the 99.6% naturally abundant 14N isotope are challenging principally due to the integer spin number (I = 1) and moderate electric quadrupole moment of the 14N nucleus. This results in 14N spectra that are anisotropically broadened by the first order quadrupolar interaction to several MHz, rendering the 14N isotope difficult to manipulate and detect with ssNMR. Accordingly, ssNMR studies typically favour the spin-1/2 nucleus 15N. The quadrupolar interaction is, however, extremely sensitive to the local structure and dynamics experienced at the 14N site, and its characterisation could provide a wealth of information not readily available from the spectra of the more commonly studied 15N isotope.1–3 One area where 14N NMR may offer significant benefits is in the field of protein ssNMR. The quadrupolar interaction experienced by each of the amide nitrogens in the peptide backbone is potentially a sensitive reporter to the backbone conformation, secondary structure and dynamics. Indeed evidence from small model peptides indicates that differences in the nature of hydrogen bonding between α-helical and β-sheets conformers can result in significant differences in the magnitude of the 14N electric field gradient (EFG) at the amide nitrogen, regardless of the type of residue.1,4 Furthermore, the analysis of natural abundance 14N in proteins opens new avenues for the study of complex medical and environmental samples that cannot be labelled for NMR studies.3
A number of strategies have been developed in order to characterise the 14N quadrupolar interaction (QI) in a variety of organic and biological materials. These include direct detection of static wideline 14N ssNMR spectra with broadband excitation methods.5–9 Alternatively, the 14N overtone transition may also be directly detected since this can potentially provide improved resolution10–14 and sensitivity15,16 since the overtone transition is unaffected by the first order quadrupolar interaction. Several methods have also been proposed for indirectly detecting 14N that employ a spin-1/2 “spy” nucleus such as 13C or 1H to indirectly detect the fundamental 14N transition in a 2D experiment.2,17–24 The indirect detection strategy is perhaps the most promising for application to complex molecules with multiple 14N sites since it benefits from the resolution and sensitivity of the “spy” nucleus, and does not suffer the low excitation bandwidth of overtone methods. We have recently reported a technique for such indirect detection that exploits moderate rf fields close to the 14N Larmor frequency, as opposed to a period of free evolution as proposed by a number of other groups,2,17–22 to generate coherence between the 14N and spy nuclei.24
In this communication we demonstrate the feasibility of recording 13C/14N correlation spectra in a full-length protein using such an indirect detection scheme, allowing the determination of the distribution of 14N quadrupolar couplings throughout the protein backbone of third IgG-binding domain of Protein G (GB3).
Dynamic nuclear polarization (DNP) offers significant improvements in sensitivity over conventional MAS-NMR,25,26 aiding many biomolecular NMR studies.27 Here we have applied high field MAS-DNP at a field of 18.8 T with sample temperatures of ca. 110 K in conjunction with 2D 13C/14N correlation spectra to enhance sensitivity and obtain spectra of a microcrystalline preparation of 13C-labelled GB3. Full experimental details are provided in the ESI.† For DNP measurements, two microcrystalline samples of GB3 were incubated with either 2 or 12.5 mM of the biradical AMUPol28 in glycerol-d8/D2O (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 v/v). In the 1H/13C cross polarization (CP) spectra of these two samples, shown in Fig. 1A and B, DNP enhancements (εon/off) of 3 and 18 were observed, respectively with relatively uniform enhancements throughout the spectrum, something mirrored in the corresponding proton-spin diffusion spectrum (ESI,† Fig. S2). Compared to spectra acquired at 273 K, a significant decrease in resolution is observed due to inhomogeneous broadening that arises upon freezing, although the resolution is favourable when compared to spectra of the homologous protein GB1 at 100 K29 (Experimental details and a discussion on the resolution observed can be found in the ESI†).
:30 v/v). In the 1H/13C cross polarization (CP) spectra of these two samples, shown in Fig. 1A and B, DNP enhancements (εon/off) of 3 and 18 were observed, respectively with relatively uniform enhancements throughout the spectrum, something mirrored in the corresponding proton-spin diffusion spectrum (ESI,† Fig. S2). Compared to spectra acquired at 273 K, a significant decrease in resolution is observed due to inhomogeneous broadening that arises upon freezing, although the resolution is favourable when compared to spectra of the homologous protein GB1 at 100 K29 (Experimental details and a discussion on the resolution observed can be found in the ESI†).
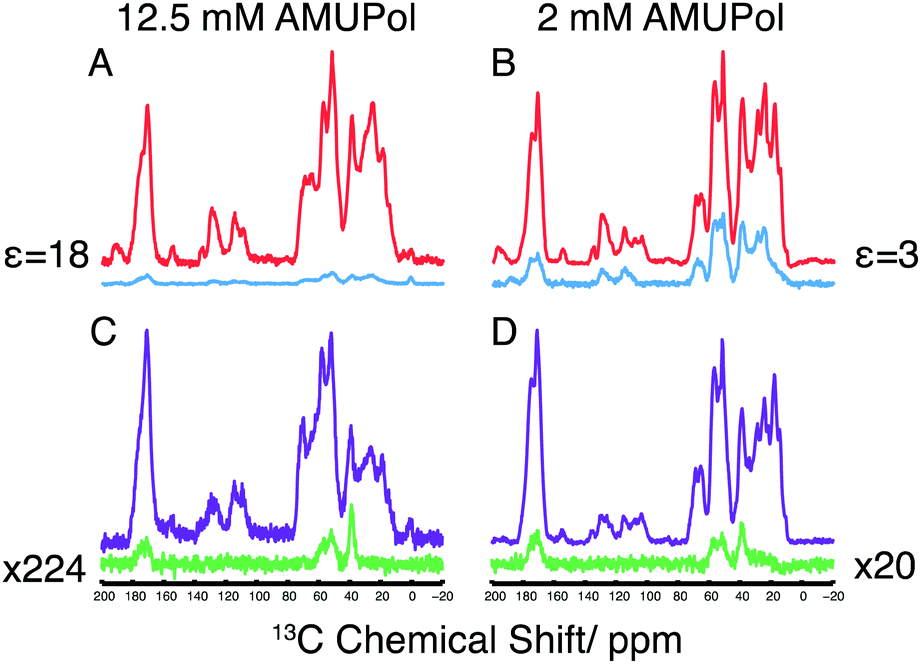 | ||
| Fig. 1 DNP enhancements and 14N transfer efficiencies recorded from ∼30 mg of GB3 in AMUPol. All spectra recorded at 18.8 T, 100 K with 13.5 kHz MAS. CP-MAS 13C NMR spectra in (A) and (B) were recorded on samples containing 12.5 mM AMUPol and 2 mM AMUPol, respectively. Spectra in red and blue show the intensity of the 13C CP-MAS with and without microwave radiation irradiation respectively. The DNP signal enhancements (ε) are indicated. Spin-echo (purple) and 14N filtered spin-echo experiments (green) recorded with DNP on the same samples containing 12.5 mM (C) and 2 mM (D) AMUPol. 14N filtered echo spectra scaling given with respect to corresponding spin-echo, with identical refocusing periods of 1.7 ms corresponding to the 14N pulse length used in both experiments. The intensity of the spectra in each panel were normalised to reflect an equivalent number of acquisitions, and where necessary, scaled by the factor shown in the figure to aid visualisation. | ||
The 14N filtered 13C spectra are shown in Fig. 1C and D. In contrast to the 1H/13C CP-MAS spectra, the 14N filtered 13C signal shows only the 13C resonances of nuclei bound to a 14N spin. Furthermore, we notice that the 14N filtered 13C spectrum is more intense at lower radical concentration, with a 10-fold improvement in signal intensity of the 2 mM AMUPol sample over the 12.5 mM AMUPol sample, when expressed as a fraction of the 13C spin echo signal under identical conditions. Typically, attenuation of the signal in the sample at high biradical concentrations is attributed to the enhanced T2′ relaxation arising from the presence of the biradicals in the sample. Here however 14N filtered intensities are compared to a 13C spin-echo signal whose refocusing periods match the duration of the 14N pulses, thereby compensating for any loss in signal due to increased 13C T2′ relaxation. This leads us to speculate that the attenuated signal observed at higher biradical concentrations arises through the enhanced relaxation of the multiple spin 14N–13C coherences during the rf driven recoupling This demonstrates the need to understand how DNP reagents influence the efficiency of different pulse sequences.30,31 To assess the feasibility of conducting these experiments in the absence of DNP, measurement have made of both microcrystalline and lyophilised material where a greater amount of protein can be packed into the rotor. In the latter case a 1D 14N filter 13C spectrum could be acquired in ∼1 week, making 2D acquisition unrealistic.
The 14N/13C 2D correlation spectra of GB3 together with the corresponding 15N/13C correlation spectra processed to mimic the inhomogeneous broadening apparent in the samples measured with DNP are shown in Fig. 2. The most intense feature in the 14N/13C correlation spectra, with 14N shifts between 280 ppm and 320 ppm, is assigned to the primary amines in the lysine sidechain on the basis of the 14N and 13C chemical shifts. In the region corresponding to the Cα and CO chemical shift (50–55 ppm and 170–175 ppm 13C shift, respectively) a broad distribution of 14N resonances are observed between 310 ppm and 420 ppm. The 14N shifts cover a range of 110 ppm, a dispersion almost four times greater than observed in the amide region of the corresponding 15N/13C spectra. Notable in their absence, are resonances with 13C chemical shifts of 44–47 ppm arising from the glycine residues in GB3. 15N/13C 2D correlation spectra (data not shown) acquired under similar DNP conditions, show little perturbation in the Cα shifts of glycine residues, whilst studies of model peptides have indicated that the 14N QI is similar to that of other amino acids. We suggest that the absence of these resonances is due to the short T2 of these residues arising from incomplete decoupling of the protons within the CH2 groups of the glycine residues with the proton decoupling fields available.
 | ||
| Fig. 2 Carbon/nitrogen correlation spectra of GB3. The top panels show the carbonyl and aliphatic regions of 13C/15N correlation spectra of U-13C,15N-GB3 recorded at 14.1 T, 293 K under 12.5 kHz MAS. Lower panels show corresponding regions of the 13C/14N correlation spectrum of U-13C GB3 containing 2 mM AMUPol recorded at 18.8 T, 100 K under 13.5 kHz MAS. | ||
The increase in 14N shift dispersion in the 14N/13C spectrum of the protein, compared to the 15N spectra, is due to the contribution of the field dependent second order isotropic quadrupolar shift (SOIQS) to the 14N shift, in addition to the usual nitrogen isotropic chemical shift. The 14N SOIQS may be given by:32
 | (1) |
 | (2) |
The observed dispersion of CQ is dependent on both the local dynamics and electronic environment at each of the amide sites in the protein backbone, the latter reflecting the conformation and H-bond status of each site. The magnitude of CQ measured agrees well with ab initio calculations4 and studies of model compounds including N-acetyl-valine,1 triglycine,33 and alanyl–glycyl–glycine17 suggesting the protein backbone is immobile with little overall dynamic averaging of the quadrupolar interaction; as expected for this well-structured protein in a glass-frozen matrix at 100 K.
The dispersion in CQ is however larger than previous studies of short model peptides, where changes from an α-helical to a β-strand conformation result in a variation of the 14N CQ of ∼200 kHz.4 The absence of any overall dynamic scaling of CQ suggests that the larger distribution in CQ reflects either smaller localised mobility leading to dynamic averaging of CQ or the greater structural diversity present in the backbone of GB3 compared to earlier studies of model compounds.
To assess whether the dispersion of 14N shifts reflects the distribution of backbone conformations present in GB3 captured here upon sample freezing at 100 K, we have modelled the 14N/13C correlation spectrum based on the SOIQS predicted from the backbone conformations in the crystal structure34 and the backbone 15N/13C assignment. Qualitatively these modelled spectra mirror the experimental data (see ESI,† Fig. S3), with the intensity in regions where the SOIQS would correlate well with α-helical and β-strands structures. This highlights the potential for such studies to provide an oversight of the secondary structures in proteins.
In conclusion, we demonstrate that combining DNP at cryogenic temperatures with 14N indirect detection it is possible to characterise the quadrupolar interactions at 14N sites within a microcrystalline preparation of GB3. Despite the enhanced linewidths observed under DNP conditions which prohibit the identification of site specific resonances, we have demonstrated the feasibility of characterising the distribution of the quadrupolar interactions through the analysis of the unresolved envelope of 14N resonances in the indirect dimension. The distribution observed reveals that the 14N sites within the protein backbone exhibit a broad range of 14N EFGs that indicate that, in the case of GB3 at these temperatures, the observed shifts reflect the secondary structures adopted by each amino acid. The exquisite sensitivity demonstrated by the EFG to the backbone conformation and the possibility to characterise these sites, offers a novel route to the structural characterisation of biomolecules.
We envision that the utility of this method will be further enhanced through the use of additional dimensions that would alleviate some of the spectra crowding arising from the DNP conditions used. Furthermore, utilization of alternative spy nuclei such as protons would further boost sensitivity whilst facilitating the NMR analysis of biomolecules and other natural products that have previously proved intractable due to difficulties associated with labelling.
This work has been supported by the EPSRC (EP/M023664/1). MC thanks the University Research Fellowship scheme from the Royal Society for support. IH thanks The Higher Committee for Education Development in Iraq for support. The authors acknowledge the use of the IRIDIS HPC Facility at the University of Southampton. Development of Spinach is supported by EPSRC (EP/H003789/1). Financial support by the Access to Research Infrastructures activity in the 7th Framework Programme of the EC (Project number: 261863, Bio-NMR) for conducting the research is gratefully acknowledged. We would like to thank Prof. Emsley and Dr Lesage for their useful discussion and access to the 800 MHz DNP spectrometer at the ISA, Lyon.
Conflicts of interest
There are no conflicts to declare.References
- R. E. Stark, R. A. Haberkorn and R. G. Griffin, J. Chem. Phys., 1978, 68, 1996 CrossRef CAS.
- S. Antonijevic and N. Halpern-Manners, Solid State Nucl. Magn. Reson., 2008, 33, 82–87 CrossRef CAS PubMed.
- G. N. Reddy, M. Malon, A. Marsh, Y. Nishiyama and S. P. Brown, Anal. Chem., 2016, 88, 11412–11419 CrossRef CAS PubMed.
- J. Fukazawa, S. Kato, T. Ozaki, A. Shoji and K. Takegoshi, J. Am. Chem. Soc., 2010, 132, 4290–4294 CrossRef CAS PubMed.
- K. J. Harris, S. L. Veinberg, C. R. Mireault, A. Lupulescu, L. Frydman and R. W. Schurko, Chemistry, 2013, 19, 16469–16475 CrossRef CAS PubMed.
- L. A. O'Dell and R. W. Schurko, J. Am. Chem. Soc., 2009, 131, 6658–6659 CrossRef PubMed.
- L. A. O'Dell and R. W. Schurko, Phys. Chem. Chem. Phys., 2009, 11, 7069–7077 RSC.
- L. A. O'Dell, R. W. Schurko, K. J. Harris, J. Autschbach and C. I. Ratcliffe, J. Am. Chem. Soc., 2011, 133, 527–546 CrossRef PubMed.
- S. L. Veinberg, Z. W. Friedl, K. J. Harris, L. A. O'Dell and R. W. Schurko, CrystEngComm, 2015, 17, 5225–5236 RSC.
- L. A. O'Dell and A. Brinkmann, J. Chem. Phys., 2013, 138, 064201 CrossRef PubMed.
- L. A. O'Dell, R. He and J. Pandohee, CrystEngComm, 2013, 15, 8657 RSC.
- L. A. O'Dell and C. I. Ratcliffe, Chem. Phys. Lett., 2011, 514, 168–173 CrossRef.
- Y. Nishiyama, M. Malon, Z. Gan, Y. Endo and T. Nemoto, J. Magn. Reson., 2013, 230, 160–164 CrossRef CAS PubMed.
- I. M. Haies, J. A. Jarvis, L. J. Brown, I. Kuprov, P. T. F. Williamson and M. Carravetta, Phys. Chem. Chem. Phys., 2015, 17, 23748–23753 RSC.
- A. J. Rossini, L. Emsley and L. A. O'Dell, Phys. Chem. Chem. Phys., 2014, 16, 12890–12899 RSC.
- I. M. Haies, J. A. Jarvis, H. Bentley, I. Heinmaa, I. Kuprov, P. T. F. Williamson and M. Carravetta, Phys. Chem. Chem. Phys., 2015, 17, 6577–6587 RSC.
- Z. H. Gan, J. Am. Chem. Soc., 2006, 128, 6040–6041 CrossRef CAS PubMed.
- Z. H. Gan, J. Magn. Reson., 2007, 184, 39–43 CrossRef CAS PubMed.
- S. Cavadini, A. Abraham and G. Bodenhausen, J. Magn. Reson., 2008, 190, 160–164 CrossRef CAS PubMed.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, ChemPhysChem, 2007, 8, 1363–1374 CrossRef CAS PubMed.
- S. Cavadini, S. Antonijevic, A. Lupulescu and G. Bodenhausen, J. Magn. Reson., 2006, 182, 168–172 CrossRef CAS PubMed.
- S. Cavadini, V. Vitzthum, S. Ulzega, A. Abraham and G. Bodenhausen, J. Magn. Reson., 2010, 202, 57–63 CrossRef CAS PubMed.
- Y. Nishiyama, Y. Endo, T. Nemoto, H. Utsumi, K. Yamauchi, K. Hioka and T. Asakura, J. Magn. Reson., 2011, 208, 44–48 CrossRef CAS PubMed.
- J. A. Jarvis, I. M. Haies, P. T. F. Williamson and M. Carravetta, Phys. Chem. Chem. Phys., 2013, 15, 7613–7620 RSC.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K. N. Hu, C. G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. van der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Chem. Phys., 2008, 128, 052211 CrossRef PubMed.
- Y. Su, L. Andreas and R. G. Griffin, Annu. Rev. Biochem., 2015, 84, 465–497 CrossRef CAS PubMed.
- C. Sauvee, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., 2013, 52, 10858–10861 CrossRef CAS PubMed.
- J. R. Lewandowski, M. E. Halse, M. Blackledge and L. Emsley, Science, 2015, 348, 578–581 CrossRef CAS PubMed.
- D. Lee, S. Hediger and G. De Paepe, Solid State Nucl. Magn. Reson., 2015, 66-67, 6–20 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, D. Gajan, F. Rascon, M. Rosay, W. E. Maas, C. Coperet, A. Lesage and L. Emsley, Chem. Sci., 2012, 3, 108–115 RSC.
- A. Samoson, Chem. Phys. Lett., 1985, 119, 29–32 CrossRef CAS.
- M. Strohmeier, D. W. Alderman and D. M. Grant, J. Magn. Reson., 2002, 155, 263–277 CrossRef CAS PubMed.
- T. S. Ulmer, B. E. Ramirez, F. Delaglio and A. Bax, J. Am. Chem. Soc., 2003, 125, 9179–9191 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed protocols for sample preparation and NMR data acquisition. Discussion on distribution of 14N shifts and resolution of sites in cryogenic DNP spectra. See DOI: 10.1039/c7cc03462h |
| ‡ Current address: Department of Life Sciences, Imperial College London, London, UK. |
| § Current address: Department of Chemistry, Mosul University, Mosul, Iraq. |
| ¶ Current address: Center for Magnetic Resonance, University of Florence, Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy. |
| || Current address: Department of Chemistry, Iowa State University, 0205 Hach Hall, 2438 Pammel Drive, Ames, US. |
| This journal is © The Royal Society of Chemistry 2017 |