
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalofunctionalization of alkenes by vanadium chloroperoxidase from Curvularia inaequalis†
Jia Jia
Dong‡
a,
Elena
Fernández-Fueyo‡
a,
Jingbo
Li
 b,
Zheng
Guo
b,
Rokus
Renirie
c,
Ron
Wever
c and
Frank
Hollmann
*a
b,
Zheng
Guo
b,
Rokus
Renirie
c,
Ron
Wever
c and
Frank
Hollmann
*a
aDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: f.hollmann@tudelft.nl; Tel: +31 (0)152 781957
bDepartment of Engineering, Aarhus University, Gustav Wieds Vej 10, 8000 Aarhus C, Denmark
cVan't Hoff Institute for Molecular Sciences (HIMS), Faculty of Science, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
First published on 22nd May 2017
Abstract
The vanadium-dependent chloroperoxidase from Curvularia inaequalis is a stable and efficient biocatalyst for the hydroxyhalogenation of a broad range of alkenes into halohydrins. Up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200000 TON with 69 s−1 TOF were observed for the biocatalyst. A bienzymatic cascade to yield epoxides as reaction products is presented.
200000 TON with 69 s−1 TOF were observed for the biocatalyst. A bienzymatic cascade to yield epoxides as reaction products is presented.
Vic-Halohydrins are valuable building blocks e.g. in natural product synthesis.1 Two functional groups endow the organic chemist with handles for further transformations such as conversion into epoxides and other fuctionalities.2 To attain vic-halohydrins, halohydroxylation of olefins appears to be the most direct approach next to ring-opening of epoxides by nucleophilic attack.3
Treatment of olefins with elementary halogens in water provides halohydrins. This approach, however, is hampered by the high reactivity of the reagents leading to side products and the corrosive reaction conditions.4 Also the formation of significant inorganic wastes renders this approach questionable from an environmental point of view. Therefore, N-halo compounds such as N-halosuccinimide represent the most commonly used reagents, or sodium periodate, despite the fact of low atom efficiency causing large amounts of waste products.5 More recently, transition metal catalysts (including W and V) have received increasing interest for the conversion of alkenes to halohydrins using H2O2 and inorganic halide as reagents.6 In addition, oxone has been successfully applied as oxidant using NH4Cl as halogen source.7
Finally, biocatalysis also in principle enables the conversion of alkenes to halohydrins using so-called haloperoxidases (E.C. 1.11.1.8, 1.11.1.10 and 1.11.1.18). These enzymes generate reactive hypohalites from the corresponding halides and hydrogen peroxide. This has been reported for example using the heme-dependent chloroperoxidase from Caldariomyces fumago (CfuCPO).8 However, the poor stability of heme-dependent haloperoxidases against H2O2 limits the broad applicability of this enzyme class and make control over the in situ concentration of H2O2 inevitable.9 In fact, the catalytic performance (in terms of turnover numbers, TON) reported for CfuCPO is too low to be of preparative value (generally a few thousand turnovers can be estimated from the publications).8c,10 Vanadium-dependent haloperoxidases are principally more robust against H2O2 and therefore maybe more practical catalysts.11
The vanadium chloroperoxidase from Curvularia inaequalis (CiVCPO) for example excels by its superb stability against H2O2 as well as demanding environmental conditions such as cosolvents and temperature.12 It was successfully applied for the catalytic halogenation of phenols13 as well as to mediate the (Aza-) Achmatowicz reaction.14
Therefore, we became interested in evaluating CiVCPO as catalyst for the halogenation of alkenes striving for more robust reactions than reported for the heme-dependent halogenases (Scheme 1).
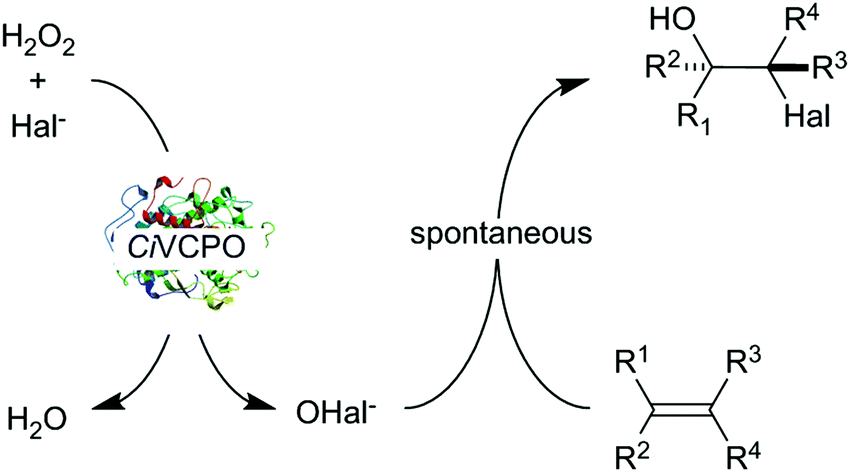 | ||
| Scheme 1 Chemoenzymatic oxidation of alkenes using the vanadium chloroperoxidase from Curvularia inaequalis (CiVCPO) as hypohalite generation catalyst. Here, the most popular chemo-enzymatic mechanism is shown wherein the enzyme forms the hypohalite in situ followed by spontaneous reaction of the latter with an alkene. It should, however be mentioned that also some indications for a direct enzymatic halohydroxylation within the enzyme active site exist.11 | ||
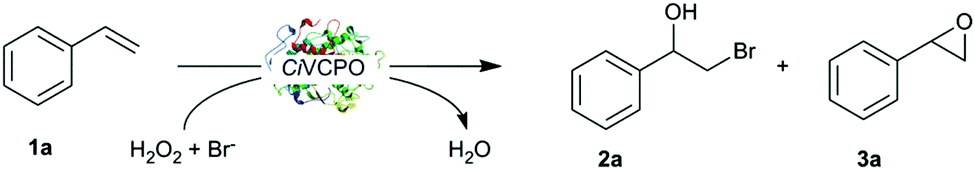
In a first set of experiments we investigated the halohydroxylation of styrene. As starting point we chose the optimised reaction conditions determined previously.13,14 Full conversion was achieved after 4 hours with high selectivity towards vic-bromohydrin (>90%) with only trace amount of side products (epoxide, ketone derivatives) detectable (Table 1, entry 2). The regio chemistry of the hydroxybromination reaction exclusively followed Markovnikov's rule. In accordance with the suspected chemo-enzymatic reaction mechanism, no enantioselectivity was observed. When the enzyme was omitted from the reaction, no product formation was observed (Table 1, entry 3), which confirms the proposed chemoenzymatic cascade. Likewise, no conversion was observed in the absence of H2O2 or KBr (data not shown).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | pHa | Solvent | KBrb | H2O2b | Conversionc (%) | Yield 2ad (%) | Yield 3ad (%) |
| a Buffers used: acetate (pH 3), citrate (pH 5) and Britton–Robinson (pH 7 and 9). b Concentration units (mM). c Determined by 1H NMR. d NMR yield determined from the crude reaction mixture. e Control reaction in the absence of CiVCPO. f 20 hours reaction time. g Portion-wise addition of H2O2. | |||||||
| 1 | 3 | H2O | 160 | 170 | 10 | 8 | n.d. |
| 2 | 5 | H2O | 160 | 170 | Full | 90 | 2 |
| 3e | 5 | H2O | 160 | 170 | 0 | 0 | 0 |
| 4f | Water | H2O | 160 | 170 | Full | 41 | 55 |
| 5f | 7 | H2O | 160 | 170 | Full | 40 | 52 |
| 6 | 9 | H2O | 160 | 170 | 13 | 4 | 8 |
| 7 | 5 | H2O | 20 | 170 | 45 | 40 | 0 |
| 8 | 5 | H2O/EtOH (2:1) |
160 | 170 | 50 | 25 | 7 |
| 9g | 5 | H2O | 160 | 110 | Full | 90 | 2 |
When using ethanol a cosolvent, also very significant amounts (13%) of (2-bromo-1-ethoxyethyl)benzene were obtained (Table 1, entry 8). We attribute this to the competition of water and ethanol as nucleophiles.
Adding the oxidant (H2O2) portion-wise proved to be more efficient than a single addition at the beginning of the reaction (Table 1, entries 2 vs. 9). This can be attributed to a the well-known formation of singlet oxygen (1O2) from hypohalites and H2O2, which in our case represents an undesired side reaction consuming H2O2.15
Varying the pH value of the reaction medium had the most significant effect on the reaction (Fig. S1, ESI†). Consistent with previous observations, highest CiVCPO activity was observed at slightly acidic to neutral conditions. Even in the absence of buffer, essentially the same results were observed as using BR (Britton–Robinson) buffer (Table 1, entries 4 and 5). Raising or lowering the pH value lead to dramatic activity decreases. Under acidic reaction conditions the bromohydrin was the predominant/sole product of the reaction. The relative epoxide yield increased with increasing pH. This is consistent with the well-known base-catalysed ring-closure of halohydrins.
To obtain further insights into the kinetics of the hydroxybromination reaction, we followed the time course of the chemoenzymatic conversion of (water soluble) sodium styrene-4-sulfonate 1g (Fig. 1). Following a very short lag-phase, the product accumulation proceeded linearly until approx. 75% conversion and then considerably slowed down. After 5–6 h, full conversion was obtained. It is worth emphasising the excellent catalytic performance of the biocatalyst here: Over 6.5 h CiVCPO performed 400000 catalytic turnovers corresponding to an average turnover frequency of more than 15 s−1 (over 6.5 h). Within the first 2 h, this value was even higher (69 s−1). Raising the concentration of 1g from 40 mM to 160 mM (while concomitantly increasing the concentration of KBr as well as the buffer strength to avoid pH shifts, vide infra) resulted in 80% of the desired product 2g. Hence CiVCPO performed at least 1280000 catalytic cycles. These values underline the robustness and efficiency of the biocatalyst.
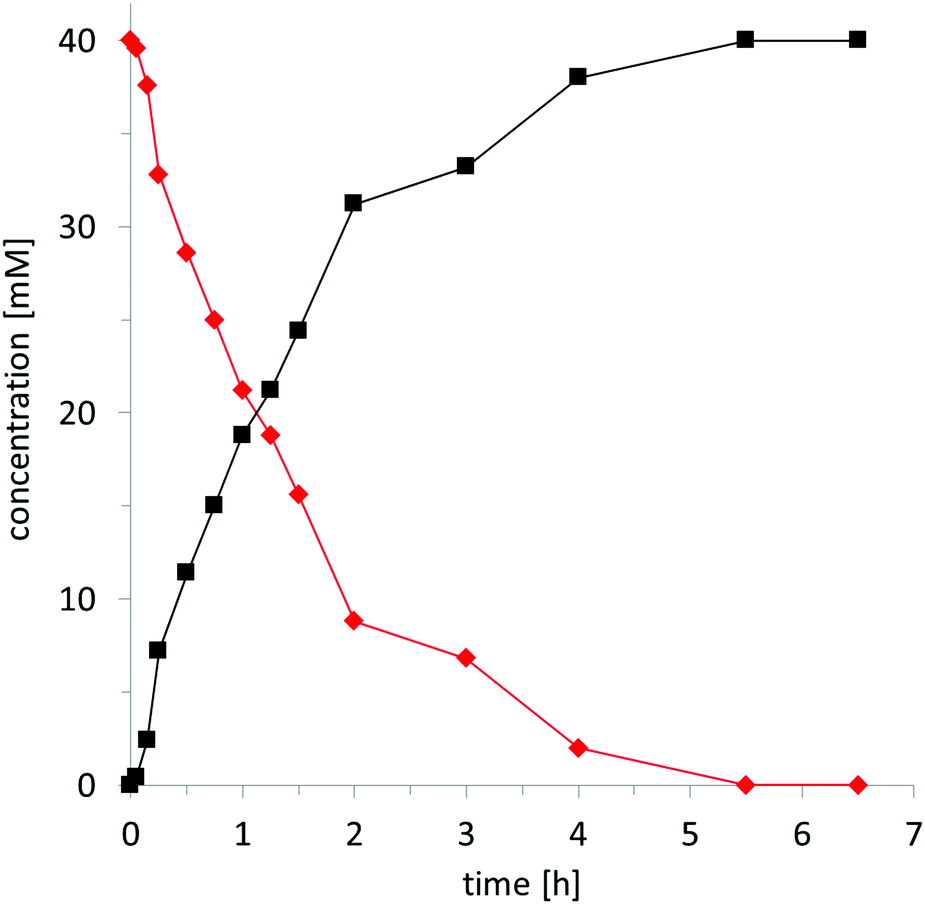 | ||
| Fig. 1 Time course of the chemoenzymatic oxidation of alkene 1g (◆) to 2g (■). General conditions: aqueous medium (D2O, 0.1 M citrate pH 5), c(1g) = 40 mM, c(KBr) = 160 mM, c(H2O2) = 170 mM, c(CiVCPO) = 100 nM, T = 25 °C. The concentration of substrate and product was determined by 1H NMR (see ESI†). | ||
Applying the optimised reaction conditions (Table 1, entry 9) we advanced to evaluate the product scope of the chemo enzymatic hydroxyhalogenation reaction. As shown in Fig. 2 both aromatic and aliphatic alkenes were converted into the corresponding halohydrins. Styrene was transformed into the corresponding bromohydrin 2a and chlorohydrin 2b with 81% and 77% yield, respectively. α- and β-methylstyrene were converted in satisfactory yields. The (E)/(Z) configuration of the latter influenced the product conformation leading to the anti-bromohydrin 2d in case of the (E)-starting material and syn-bromohydrin 2e from the (Z)-substrate. Allylbenzene 1f shows lower reactivity, yielding 51% bromohydrin 2f at 63% conversion. Exchanging bromide by chloride generally lead to a slight reduction in yields with the exception of 4-styrenesulfonic acid 1h where next to the desired chlorohydrin 2h also approx. 10% of 4-formylbenzenesulfonate were observed (Fig. 2).
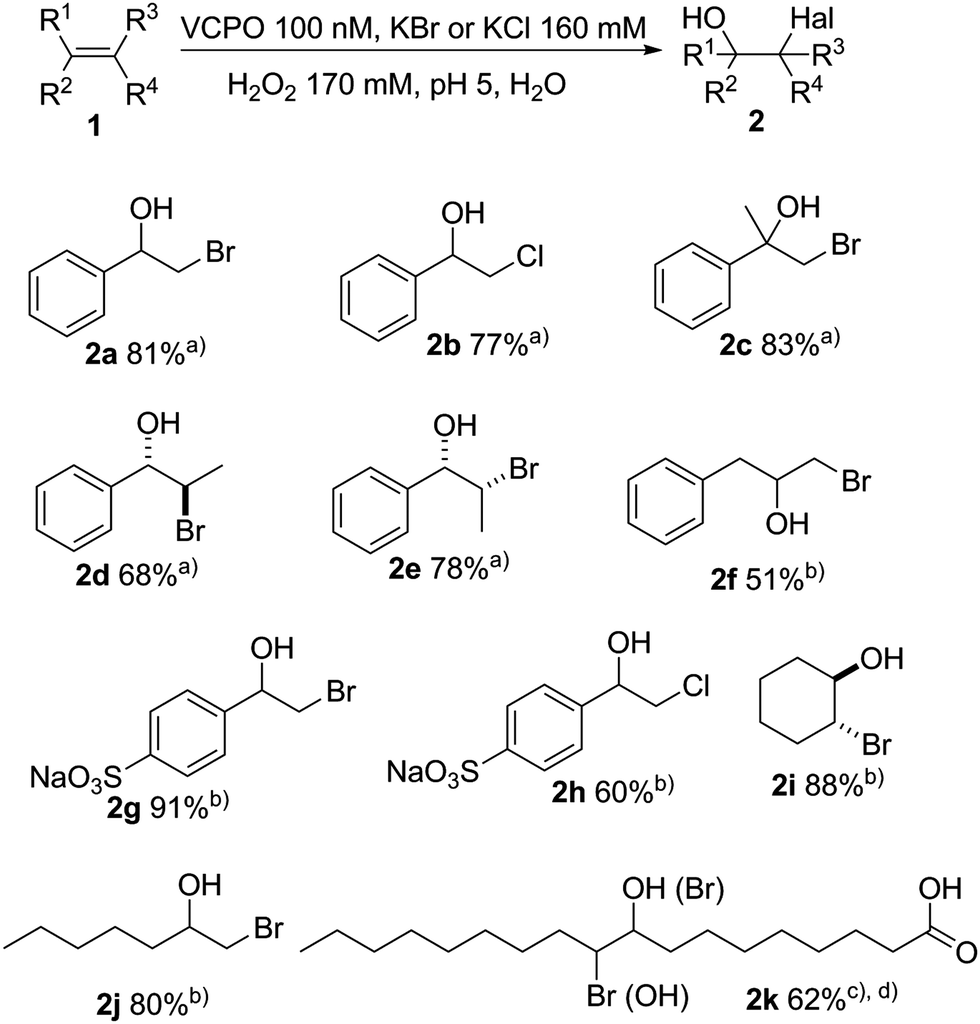 | ||
| Fig. 2 Preliminary substrate scope of the chemoenzymatic hydroxyl halogenation scheme. Reaction condition: c(CiVCPO) = 100 nM, c(alkene) = 40 mM, c(KX) = 160 mM, c(H2O2) = 170 mM, citrate buffer (0.1 M, pH 5), T = 25 °C, t = 20 h. (a) Isolated yield; (b) 1H NMR yield. (c) Ethanol as co-solvent; (d) yield of two regioisomers (see ESI†). | ||
Likewise, aliphatic alkenes were also converted smoothly: 2-bromocyclohexan-1-ol 2i and 1-bromoheptan-2-ol 2j were obtained in good yields (88% and 80%, respectively). Due to its hydrophobicity, oleic acid required some cosolvent to enhance its solubility enabling an 62% yield of the bromohydrin 2k. The lower yield is due to the 10(9)-bromo-9(10)-ethoxyoctadecanoic acid formation as side product as ethanol competed as nucleophile (Fig. 2).
Preparative scale (1.1 gram) reactions of some selected alkenes were performed. For example styrene 1a was converted almost quantitatively albeit at lower selectivity than shown in Fig. 2. After 24h the desired bromohydrin was isolated in only 50% yield whereas styrene oxide accumulated to overall 30%. We attribute this to the prolonged reaction time and an alkaline shift of the reaction pH value favouring the ring-closure reaction. Future experiments with pH control are likely to circumvent this current limitation.
Nevertheless, we became interested in the further transformation of the halohydrins into epoxides. The pH optimum of CiVCPO within the acidic range does not allow for efficient concurrent hydroxyhalogenation and ring closure. This may be circumvented by a two-step procedure wherein the halohydrin formation reaction is performed first at acidic pH followed by a base treatment to pH 10 with NaOH. This method proved to be successful for most products with the exception of the halohydrin obtained from oleic acid (2k). Here, intensive foaming (of the carboxylate) interfered with product isolation, which could be avoided by substituting NaOH with trimethylamine. Also, it is worth mentioning here that the previously mentioned 10(9)-bromo-9(10)-ethoxyoctadecanoic acid was also retained here (approx. 30% yield).
Even though this two-step procedure was efficient for the production of epoxides, we envision a more elegant (and less wasteful) approach. For this, we evaluated a bienzymatic cascade reaction comprising CiVCPO-initiated halohydrin formation followed by halohydrin dehalogenase (HHe)-catalysed ring-closure (Scheme 2). Such a cascade would, in principle, also allow for catalytic use of the halides. For this, we recombinantly expressed the HHe from Agrobacterium radiobacter. Particularly, its poor enantioselectivity appeared attractive in this case as a more enantioselective version would lead to kinetic resolution of the racemic bromohydrin and consequently in decreased yields of the desired epoxide.16
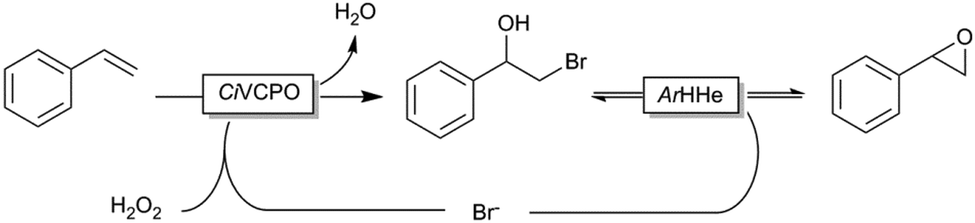 | ||
| Scheme 2 Envisioned cascade for the direct transformation of alkenes (e.g. styrene) into epoxides by combining the CiVCPO-initiated halohydrin formation to ring-closure catalysed by a halohydrin dehalogenase (e.g. from Agrobacterium radiobacter, ArHHe). | ||
A reaction employing 20 mM styrene 1a in the presence of 5 mM KBr indeed gave 6.6 mM of the desired epoxide and 1.8 mM of the bromohydrin indicating the principal feasibility of the reaction shown in Scheme 2, despite the reduced activity of biocatalysts at pH 7 (Fig. S2, ESI†).
The present study underlines the synthetic potential of the vanadium-dependent chloroperoxidase from Curvularia inaequalis. Its high robustness and activity enabled efficient halohydroxylation of various alkenes to the corresponding halohydrins on preparative scale. Despite the promising first results, a range of issues will have to be overcome to render this method truly practical. First, efficient in situ H2O2 generation systems17 will be applied to this reaction in order to circumvent the undesired 1O2 formation reaction. Also the envisioned bienzymatic cascade forming epoxides from alkenes (while using catalytic amounts of halides) will need further improvements such as the application of CiVCPO mutants with engineered pH optima,18 reaction engineering approaches utilizing spatially separated reaction compartments and immobilized enzymes.19
This study was funded by The Netherlands Organisation for Scientific Research (NWO) through a VICI grant (no. 724.014.003).
Notes and references
- G. W. Gribble, Chemosphere, 2003, 52, 289–297 CrossRef CAS PubMed.
- (a) T. Benaïssa, S. Hamman and C. G. Beguin, J. Fluorine Chem., 1988, 38, 163–173 CrossRef; (b) J. T. Tang, J. L. Zhu, Z. X. Shen and Y. W. Zhang, Tetrahedron Lett., 2007, 48, 1919–1921 CrossRef CAS; (c) H. H. Xu and C. Wolf, Angew. Chem., Int. Ed., 2011, 50, 12249–12252 CrossRef CAS PubMed; (d) O. A. Davis and J. A. Bull, Angew. Chem., Int. Ed., 2014, 53, 14230–14234 CrossRef CAS PubMed.
- (a) M. A. Reddy, K. Surendra, N. Bhanumathi and K. R. Rao, Tetrahedron, 2002, 58, 6003–6008 CrossRef CAS; (b) V. S. C. de Andrade and M. C. S. de Mattos, Synthesis, 2016, 1381–1388 CAS; (c) A. Castellanos and S. P. Fletcher, Chem. – Eur. J., 2011, 17, 5766–5776 CrossRef CAS PubMed; (d) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS PubMed.
- (a) R. E. Buckles and J. E. Maurer, J. Org. Chem., 1953, 18, 1585–1590 CrossRef; (b) S. Arai, T. Takeuchi, M. Ishikawa, T. Takeuchi, M. Yamazaki and M. Hida, J. Chem. Soc., Perkin Trans. 1, 1987, 481–487 RSC; (c) G. Y. T. Liu, W. F. Richey, J. E. Betso, B. Hughes, J. Klapacz and J. Lindner, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 DOI:10.1002/14356007.a06_565.pub2.
- (a) P. Phukan, P. Chakraborty and D. Kataki, J. Org. Chem., 2006, 71, 7533–7537 CrossRef CAS PubMed; (b) L. S. de Almeida, P. M. Esteves and M. C. S. de Mattos, Synlett, 2006, 1515–1518 CAS; (c) M. Narender, M. S. Reddy, Y. V. D. Nageswar and K. R. Rao, J. Mol. Catal. A: Chem., 2006, 258, 10–14 CrossRef CAS; (d) B. Das, K. Venkateswarlu, K. Damodar and K. Suneel, J. Mol. Catal. A: Chem., 2007, 269, 17–21 CrossRef CAS; (e) X. Q. Yang, J. Wu, X. W. Mao, T. F. Jamison and T. A. Hatton, Chem. Commun., 2014, 50, 3245–3248 RSC; (f) M. H. Shinde and U. A. Kshirsagar, Org. Biomol. Chem., 2016, 14, 858–861 RSC; (g) G. K. Dewkar, S. V. Narina and A. Sudalai, Org. Lett., 2003, 5, 4501–4504 CrossRef CAS PubMed; (h) J. Zhang, J. Wang, Z. Qiu and Y. Wang, Tetrahedron, 2011, 67, 6859–6867 CrossRef CAS.
- (a) A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef PubMed; (b) V. Conte and B. Floris, Inorg. Chim. Acta, 2010, 363, 1935–1946 CrossRef CAS; (c) T. Moriuchi, M. Yamaguchi, K. Kikushima and T. Hirao, Tetrahedron Lett., 2007, 48, 2667–2670 CrossRef CAS; (d) K. Kikushima, T. Moriuchi and T. Hirao, Chem. – Asian J., 2009, 4, 1213–1216 CrossRef CAS PubMed; (e) B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350–8359 CrossRef CAS PubMed; (f) S. Lopez, L. Rondot, C. Cavazza, M. Iannello, E. Boeri-Erba, N. Burzlaff, F. Strinitz, A. Jorge-Robin, C. Marchi-Delapierre and S. Menage, Chem. Commun., 2017, 53, 3579–3582 RSC.
- P. Swamy, M. A. Kumar, M. M. Reddy, M. Naresh, K. Srujana and N. Narender, RSC Adv., 2014, 4, 26288–26294 RSC.
- (a) Y. Liu, Y. Wang, Y. Jiang, M. Hu, S. Li and Q. Zhai, Biotechnol. Prog., 2015, 31, 724–729 CrossRef CAS PubMed; (b) J. Y. Wu, C. Liu, Y. C. Jiang, M. C. Hu, S. N. Li and Q. G. Zhai, Catal. Commun., 2010, 11, 727–731 CrossRef CAS; (c) B. A. Kaup, U. Piantini, M. Wust and J. Schrader, Appl. Microbiol. Biotechnol., 2007, 73, 1087–1096 CrossRef CAS PubMed; (d) F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef CAS PubMed; (e) J. Geigert, S. L. Neidleman, D. J. Dalietos and S. K. Dewitt, Appl. Environ. Microbiol., 1983, 45, 366–374 CAS; (f) J. Geigert, T. D. Lee, D. J. Dalietos, D. S. Hirano and S. L. Neidleman, Biochem. Biophys. Res. Commun., 1986, 136, 778–782 CrossRef CAS PubMed.
- (a) S. Bormann, A. Gomez Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 RSC; (b) J. Naapuri, J. D. Rolfes, J. Keil, C. Manzuna Sapu and J. Deska, Green Chem., 2017, 19, 447–452 RSC.
- (a) S. Aoun and M. Baboulene, J. Mol. Catal. B: Enzym., 1998, 4, 101–109 CrossRef CAS; (b) K. Ramakrishnan, M. E. Oppenhuizen, S. Saunders and J. Fisher, Biochemistry, 1983, 22, 3271–3277 CrossRef CAS PubMed.
- (a) H. Yamada, N. Itoh and Y. Izumi, J. Biol. Chem., 1985, 260, 1962–1969 Search PubMed; (b) N. Itoh, L. Y. Cheng, Y. Izumi and H. Yamada, J. Biotechnol., 1987, 5, 29–38 CrossRef CAS; (c) N. Itoh, A. Hasan, Y. Izumi and H. Yamada, Eur. J. Biochem., 1988, 172, 477–484 CrossRef CAS PubMed; (d) F. Natalio, S. Wiese, W. Brandt and L. Wessjohann, Chem. – Eur. J., 2017, 23, 4973–4980 CrossRef CAS PubMed; (e) A. Frank, C. J. Seel, M. Groll and T. Gulder, ChemBioChem, 2016, 17, 2028–2032 CrossRef CAS PubMed; (f) C. Leblanc, H. Vilter, J. B. Fournier, L. Delage, P. Potin, E. Rebuffet, G. Michel, P. L. Solari, M. C. Feiters and M. Czjzek, Coord. Chem. Rev., 2015, 301–302, 134–146 CrossRef CAS; (g) D. Wischang, M. Radlow and J. Hartung, Dalton Trans., 2013, 42, 11926–11940 RSC; (h) D. Wischang and J. Hartung, Tetrahedron, 2012, 68, 9456–9463 CrossRef CAS; (i) R. Wever, Vanadium: Biochemical and Molecular Biological Approaches, Springer, 2012, pp. 95–125 Search PubMed; (j) D. Wischang and J. Hartung, Tetrahedron, 2011, 67, 4048–4054 CrossRef CAS; (k) D. Wischang, O. Brucher and J. Hartung, Coord. Chem. Rev., 2011, 255, 2204–2217 CrossRef CAS; (l) J. N. Carter-Franklin and A. Butler, J. Am. Chem. Soc., 2004, 126, 15060–15066 CrossRef CAS PubMed; (m) A. Butler and M. Sandy, Nature, 2009, 460, 848–854 CrossRef CAS PubMed; (n) R. Gupta, G. J. Hou, R. Renirie, R. Wever and T. Polenova, J. Am. Chem. Soc., 2015, 137, 5618–5628 CrossRef CAS PubMed; (o) S. Diethelm, R. Teufel, L. Kaysser and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11023–11026 CrossRef CAS PubMed.
- (a) H. B. ten Brink, H. L. Dekker, H. E. Shoemaker and R. Wever, J. Inorg. Biochem., 2000, 80, 91–98 CrossRef CAS PubMed; (b) W. Hemrika, R. Renirie, S. Macedo-Ribeiro, A. Messerschmidt and R. Wever, J. Biol. Chem., 1999, 274, 23820–23827 CrossRef CAS PubMed; (c) A. Messerschmidt, L. Prade and R. Wever, Biol. Chem., 1997, 378, 309–315 CrossRef CAS PubMed.
- E. Fernández-Fueyo, M. van Wingerden, R. Renirie, R. Wever, Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2015, 7, 4035–4038 CrossRef.
- E. Fernández-Fueyo, S. H. H. Younes, S. V. Rootselaar, R. W. M. Aben, R. Renirie, R. Wever, D. Holtmann, F. P. J. T. Rutjes and F. Hollmann, ACS Catal., 2016, 6, 5904–5907 CrossRef.
- R. Renirie, C. Pierlot, J.-M. Aubry, A. F. Hartog, H. E. Schoemaker, P. L. Alsters and R. Wever, Adv. Synth. Catal., 2003, 345, 849–858 CrossRef CAS.
- (a) R. M. Haak, F. Berthiol, T. Jerphagnon, A. J. A. Gayet, C. Tarabiono, C. P. Postema, V. Ritleng, M. Pfeffer, D. B. Janssen, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2008, 130, 13508–13509 CrossRef CAS PubMed; (b) R. M. Haak, C. Tarabiono, D. B. Janssen, A. J. Minnaard, J. G. de Vries and B. L. Feringa, Org. Biomol. Chem., 2007, 5, 318–323 RSC; (c) A. Schallmey and M. Schallmey, Appl. Microbiol. Biotechnol., 2016, 100, 7827–7839 CrossRef CAS PubMed; (d) M. Schallmey, R. J. Floor, B. Hauer, M. Breuer, P. A. Jekel, H. J. Wijma, B. W. Dijkstra and D. B. Janssen, ChemBioChem, 2013, 14, 870–881 CrossRef CAS PubMed.
- (a) D. S. Choi, Y. Ni, E. Fernández-Fueyo, M. Lee, F. Hollmann and C. B. Park, ACS Catal., 2017, 7, 1563–1567 CrossRef CAS; (b) Y. Ni, E. Fernández-Fueyo, A. G. Baraibar, R. Ullrich, M. Hofrichter, H. Yanase, M. Alcalde, W. J. H. van Berkel and F. Hollmann, Angew. Chem., Int. Ed., 2016, 55, 798–801 CrossRef CAS PubMed; (c) E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 CrossRef CAS PubMed; (d) D. I. Perez, M. Mifsud Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 CAS; (e) D. Holtmann, T. Krieg, L. Getrey and J. Schrader, Catal. Commun., 2014, 51, 82–85 CrossRef CAS; (f) T. Krieg, S. Huttmann, K.-M. Mangold, J. Schrader and D. Holtmann, Green Chem., 2011, 13, 2686–2689 RSC; (g) S. Lutz, E. Steckhan and A. Liese, Electrochem. Commun., 2004, 6, 583–587 CrossRef CAS.
- Z. Hasan, R. Renirie, R. Kerkman, H. J. Ruijssenaars, A. F. Hartog and R. Wever, J. Biol. Chem., 2006, 281, 9738–9744 CrossRef CAS PubMed.
- (a) U. Hanefeld, L. Gardossi and E. Manger, Chem. Soc. Rev., 2009, 38, 453–468 RSC; (b) R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC; (c) R. Dicosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC; (d) C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures catalytic studies, spectra, enzyme preparation (CiVCPO, HHe). See DOI: 10.1039/c7cc03368k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |