
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidation of the elusive structure and formula of the active pharmaceutical ingredient bismuth subgallate by continuous rotation electron diffraction†
Yunchen
Wang
a,
Sofia
Takki
a,
Ocean
Cheung
b,
Hongyi
Xu
a,
Wei
Wan
a,
Lars
Öhrström
 c and
A. Ken
Inge
*a
c and
A. Ken
Inge
*a
aDepartment of Material and Environmental Chemistry, Stockholm University, 106 91 Stockholm, Sweden. E-mail: andrew.inge@mmk.su.se
bSchool of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden
First published on 7th June 2017
Abstract
Bismuth subgallate has been used in wound and gastrointestinal therapy for over a century. The combination of continuous rotation electron diffraction and sample cooling finally revealed its structure as a coordination polymer. The structure provides insight regarding its formula, poor solubility, acid resistance and previously unreported gas sorption properties.
When the active pharmaceutical ingredient (API) in a drug has both an unknown structure and unclear formula, an unsatisfactory situation results for all stakeholders, from patients to regulatory bodies. Structure determination is paramount for understanding physical and chemical properties such as solubility, which the pharmaceutical properties are often critically dependent on. However, structure determination is unfortunately not always trivial. Complications such as crystal size, poor stability during characterization, and complexity of the structures can impede structure investigations by conventional techniques. We now resolve this situation for bismuth(III) gallate (bismuth subgallate) using a combination of continuous rotation electron diffraction and sample cooling. The compound has been used in medicine for over a century to treat wounds, gastrointestinal disorders and syphilis.1 Today it is the API of numerous over-the-counter drugs including Devrom®, Stryphnasal® N and Sulbogin® (Suile™).2 In addition to efficacy as an antimicrobial agent, drugs containing the title compound are also used as astringents, haemostatic agents, and internal deodorants.3 Until now, due to lack of structural evidence, it has been presented as a molecular compound, either with Bi3+ chelated by two deprotonated phenolate groups, one phenol group intact and a coordinated hydroxyl ion, or with Bi3+ coordinated by a carboxylate group and two hydroxyl ions (i.e. in the US pharmacopeia, Chemspider database etc.). However, we have discovered that both structure and formula are actually somewhat different from earlier proposals.
While bismuth compounds have historically been used in cosmetics and in medicine for over three centuries,4 today much focus is on their applications in treating Helicobacter pylori infections due to their implications on society – at least half of the world's population is infected with H. pylori.5 These infections are linked to peptic and duodenal ulcers, gastritis and stomach cancer.6 Orally administered bismuth compounds have been reported to form crystalline coatings over ulcers craters hastening healing.7 Antibiotic resistance is a growing threat to global health, and it has been demonstrated that the co-administration of bismuth compounds with organic antibiotics and proton-pump inhibitors are effective in the treatment of antibiotic resistant strains of H. pylori.8 Structure determination of bismuth compounds used as APIs7a,9 has been impeded by their tendency to form as nanocrystalline powders rather than large crystals which prevents structure determination by the conventional method of choice: single crystal X-ray diffraction. Additionally, their pronounced tendency of oligomerization results in complex species such as [Bi38O44(Hsal)26(Me2CO)16(H2O)2]·(Me2CO)4,9d which complicates structure determination by X-ray powder diffraction (XPD). Moreover, variations in pH may have a large impact.10 Consequently, despite their current and historical applications in medicine, details of their structures are just emerging.7a,9d,e Obviously, this lack of structural information is linked to the still sketchy knowledge of the mode of action of these drugs. Therefore it is paramount to develop and implement new structure characterization methods in order to overcome issues preventing crystal structure elucidation.
The structure determination of bismuth subgallate required recently developed crystallographic techniques, as all attempts by conventional methods failed. Single crystal X-ray diffraction was not plausible due to small crystal size (ca. 6 × 4 × 0.2 μm, Fig. S1, ESI†). From XPD data (Fig. S2, ESI†) unit cell parameters, several possible space groups, and Bi3+ positions were determined, however efforts to locate any other atom in the structure were unsuccessful. We then turned to electron diffraction (ED),11 where 3D data can be collected from nano-sized single crystals in a transmission electron microscope (TEM).12 However, a major limitation with crystalline organic or metal–organic compounds is their tendency to be quickly destroyed under the high-energy electron beam. Rotation electron diffraction (RED)11b,c data were initially collected under ambient temperature. Beam damage, poor resolution and significant diffuse scattering were observed in all data sets (Fig. 1a and c), and not even the unit cell parameters could be confirmed with this data. Data collection was reattempted, but with two significant changes in an effort to acquire better quality diffraction data. Firstly, continuous rotation data collection was performed by acquiring electron diffraction frames on a high-speed hybrid detection camera (Timepix Quad) in video mode following the method described by Gemmi et al. In this method the sample is continuously rotated throughout the entire data acquisition process.13 This significantly reduced the data collection time from one hour to three minutes. Secondly, the sample was cooled to 173 K on a cryo-transfer sample holder before insertion into the microscope, and further cooled down to 98 K before data collection. As a result of these two actions, beam damage was minimized, the resolution of the data was greatly improved, and no obvious diffuse scattering was observed (Fig. 1b and d and Fig. S3, ESI†) in agreement with the XPD data (Fig. S2 and S4, ESI†). This suggests that disorder, as indicated by the presence of diffuse scattering in earlier data sets, was not present in the original sample, but was introduced in the TEM without sample cooling and fast data collection. Metal–organic frameworks14 and coordination polymers15 can exhibit diffuse scattering as a result of the degradation of long-range order upon removal of solvent molecules.
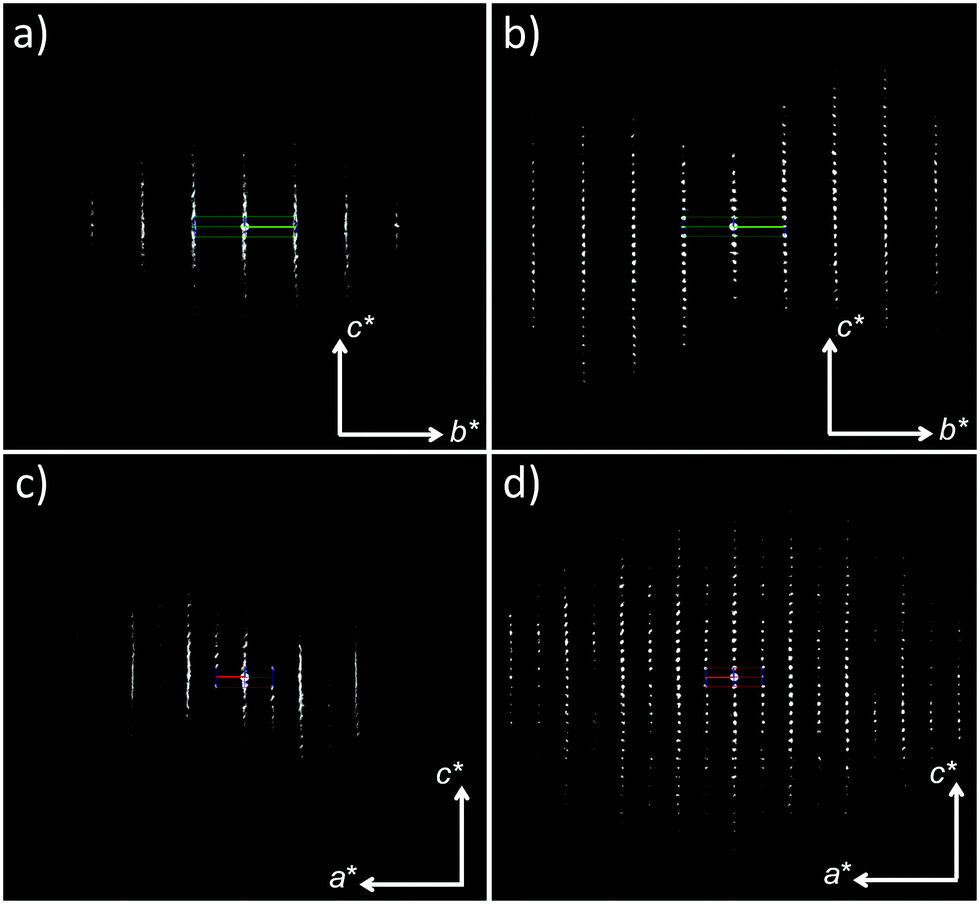 | ||
| Fig. 1 Electron diffraction data collected on bismuth subgallate by (a and c) conventional RED and (b and d) continuous rotation electron diffraction with sample cooling. | ||
The new ED data indicated similar unit cell parameters as determined by XPD and the extinction symbol, P-na, was unambiguously determined from the reflection conditions. Structure determination was performed in the space group Pmna using the ED data and to our surprise the positions of all non-hydrogen atoms in the crystal structure, including those of water molecules in the pores (Fig. S5, ESI†), were determined in the initial structure solution. Due to high R-values as a consequence of dynamical scattering common to electron diffraction data12a and low data completeness, Rietveld refinement was performed using XPD data (Fig. S4, ESI†).‡
Crystal structure determination revealed that this API is in fact a coordination polymer.15 All three phenolate oxygen atoms of the gallate ligand coordinate to Bi3+, while the carboxylic acid group does not (Fig. 2). The phenolate oxygens in the 3- and 5-positions coordinate to separate Bi3+. The phenolate oxygen in the 4-position bridges these two Bi3+. Each Bi3+ is coordinated by two gallate ligands resulting in [–Bi–O–]∞ rods parallel to the a-axis (Fig. 1b). The rods appear V-shaped when viewed down their lengths due to the positioning of the protruding ligands (Fig. 1d). Additionally, one water molecule also bridges every pair of adjacent Bi3+. The coordination geometry around Bi3+ is that of a severely distorted octahedron.
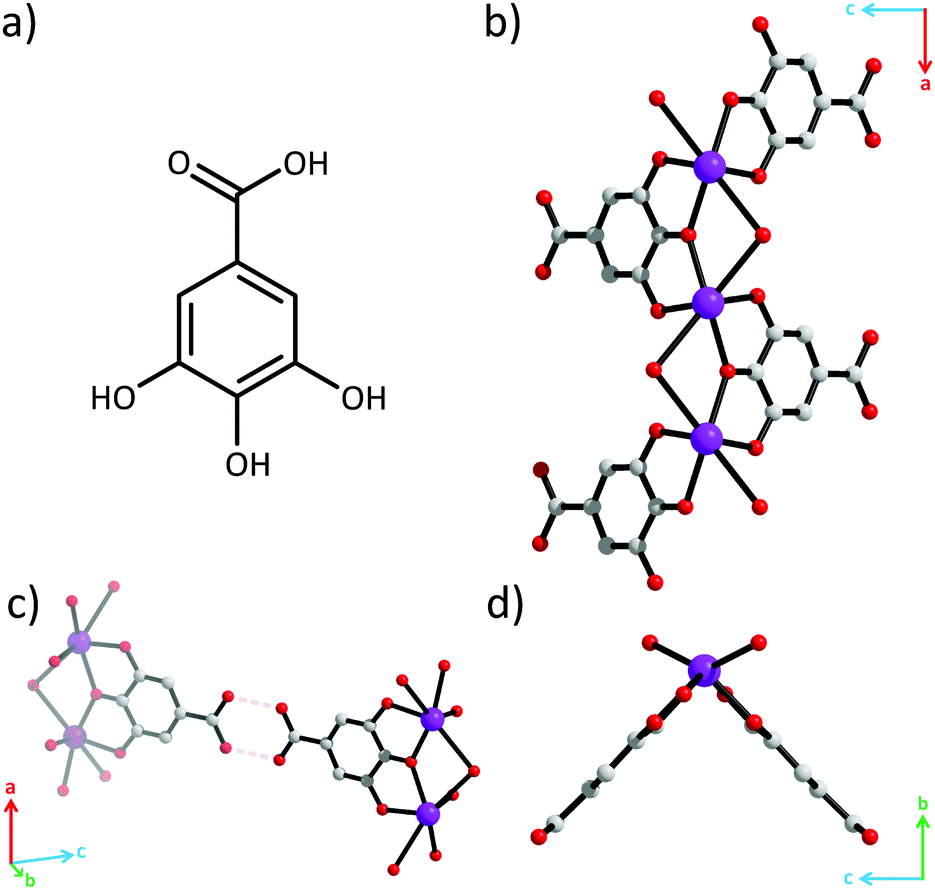 | ||
| Fig. 2 (a) Gallic acid. (b) [–Bi–O–]∞ rods in bismuth subgallate. Bismuth, carbon and oxygen atoms are represented as purple, grey and red spheres. Hydrogen atoms are omitted for clarity. (c) Hydrogen bonds between carboxylic acids. (d) Rods viewed down the a-axis. | ||
Although hydrogen atoms were not located in the difference Fourier maps it is evident from the classical hydrogen bond pattern that the carboxylic acid remains protonated. The carboxylic acid groups protrude out perpendicular to the lengths of the rods and form hydrogen bonds with carboxylic acid groups on neighbouring rods with O(H)⋯O distances of 2.82(3) Å and CO(H)⋯O angles of 119(1)° (Fig. 2c and Fig. S6, ESI†). This classical hydrogen bond motif holds the rods together into zigzag-shaped layers (Fig. 3). The layers stack resulting in channels parallel with the b-axis.
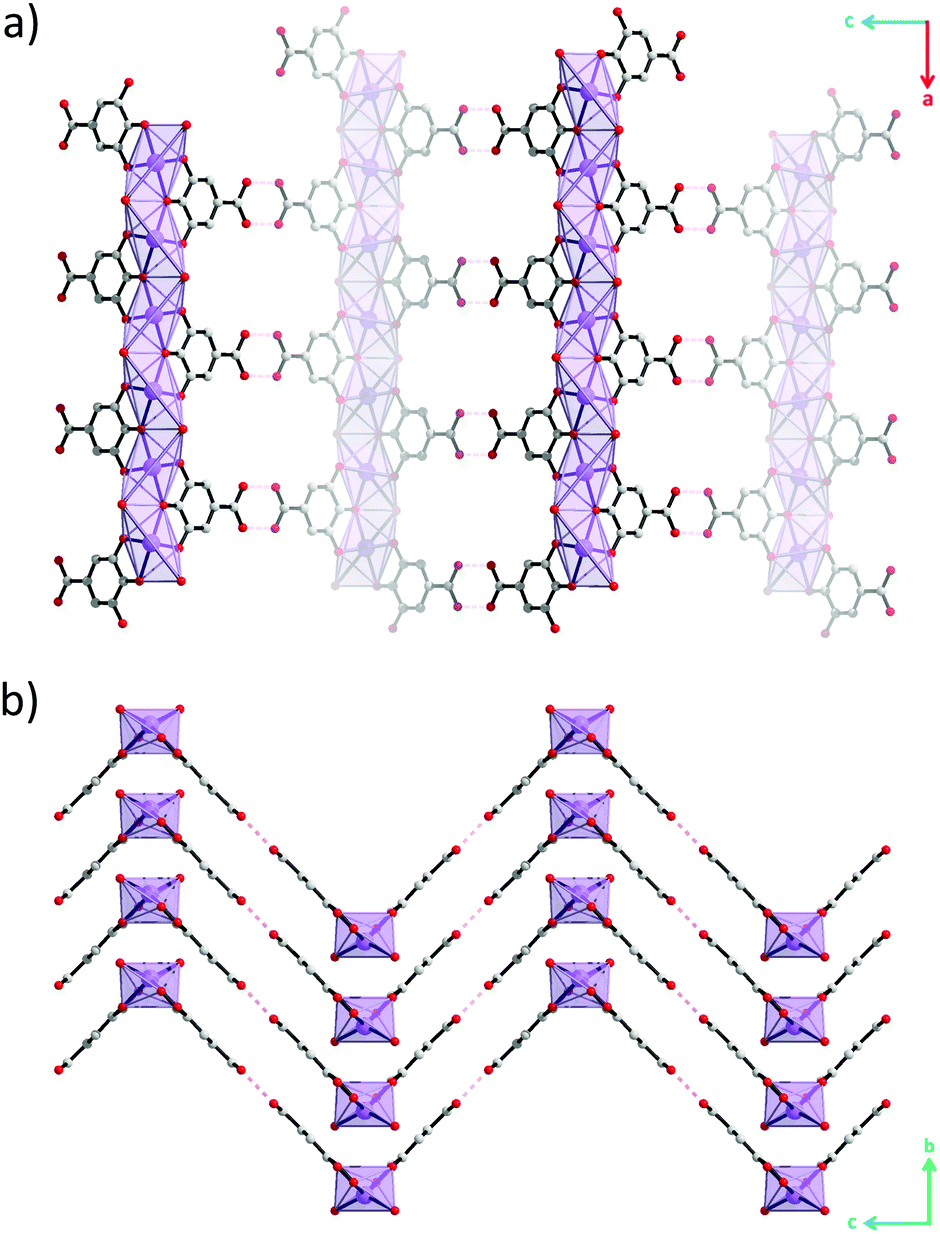 | ||
| Fig. 3 Crystal structure of bismuth subgallate viewed along (a) [010] and (b) [100]. Bismuth, carbon and oxygen atoms are coloured purple, grey and red respectively. Hydrogen atoms and water molecules in the pores have been omitted for clarity. | ||
Two water molecules per asymmetric unit were located in the pores forming hydrogen bonds with one another as well as with the carboxylic acid and the bridging water molecule in the rods (Fig. S5, ESI†). The pore volume was determined by the SQUEEZE routine in PLATON16 as 45 Å3 per Bi3+ which suggests that fewer than three water molecules can reside in the pores, assuming non-hydrogen atoms occupy an estimated volume of 18 Å3.
We propose that charge balance of Bi3+ is achieved through deprotonation of the three phenolate groups. Bi–O distances of 2.03(2), 2.227(8), and 2.03(2) Å are observed (Fig. S6, ESI†) which are more consistent with Bi3+–O− distances in structures where two Bi3+ are bridged by a deprotonated phenolate group (ca. 2.2 Å),17 rather than the longer protonated Bi3+–OH phenol bridge (ca. 2.6 Å).18 The rather long Bi3+–OH2 distance of 2.83(2) Å observed in the title compound is more consistent with typical bond lengths of water bridging two Bi3+ (ca. 2.8 Å)17b,19 rather than hydroxyl groups (ca. 2.2 Å).20
Thus we propose that the chemical composition of bismuth subgallate should be formulated as [Bi(C6H2(O)3COOH)(H2O)]n·2nH2O.
Thermogravimetric analysis performed in air (Fig. S7, ESI†) indicated a weight loss of 11% (calc. 12%) between 30–150 °C, attributed to the loss of the three water molecules per Bi3+ in agreement with previous reports.21 However we attribute the loss as two water molecules from the pores and the third as the bridging water molecule in the rods. Subsequent loss of the organic ligand occurred between 260–300 °C.
The X-ray thermodiffraction data (Fig. S8, ESI†) indicate a partial degradation in crystallinity starting at 150 °C, coinciding with the removal of the water molecules. The material becomes amorphous at 300 °C. Heating a pristine sample in air to 175 °C followed by cooling back down to 50 °C and leaving it in air for one hour allowed full crystallinity to be recovered (Fig. S9 and S10, ESI†). Alternatively, adding a drop of water immediately restores full crystallinity. The crystal structure of bismuth subgallate was found to remain intact after stirring samples in aqueous HCl solutions at 37 °C (Fig. S11, ESI†) indicating stability under acidic conditions similar to that of gastric acid (pH 1.5–3.5). The poor solubility and good stability of the material in acidic media are attributed to the extended structure and the strong Bi3+–O(phenolate)− bonds.
Bismuth compounds tend to crystallize as dense phases and we are only aware of a few porous bismuth coordination polymers.22 The N2 adsorption isotherm of bismuth subgallate recorded at 78 K showed an IUPAC Type II isotherm indicative of a non-porous material (Fig. S12, ESI†).23 However, at 273–293 K noticeable uptake of both CO2 and N2 was detected, with high selectivity towards CO2 (Fig. 4 and Fig. S13–S15, ESI†). The uptake of both gases unexpectedly increased when the temperature was raised from 273 to 283 K (CO2: from 0.94 mmol g−1 to 1.04 mmol g−1, N2: 0.10 mmol g−1 to 0.18 mmol g−1, at 101 kPa). As adsorption is an exothermic process, such increase in gas uptake with increasing temperature was unexpected. Both the uptake of CO2 and N2 reduced when the temperature was further increased from 283 to 293 K (CO2: 1.01 mmol g−1 N2: 0.16 mmol g−1, at 101 kPa). The unexpected increase in gas uptake at 283 K suggests that the gas accessible voids of bismuth subgallate are temperature dependent, which implies that the structure of bismuth subgallate is slightly expanding or rearranging with varying temperatures.
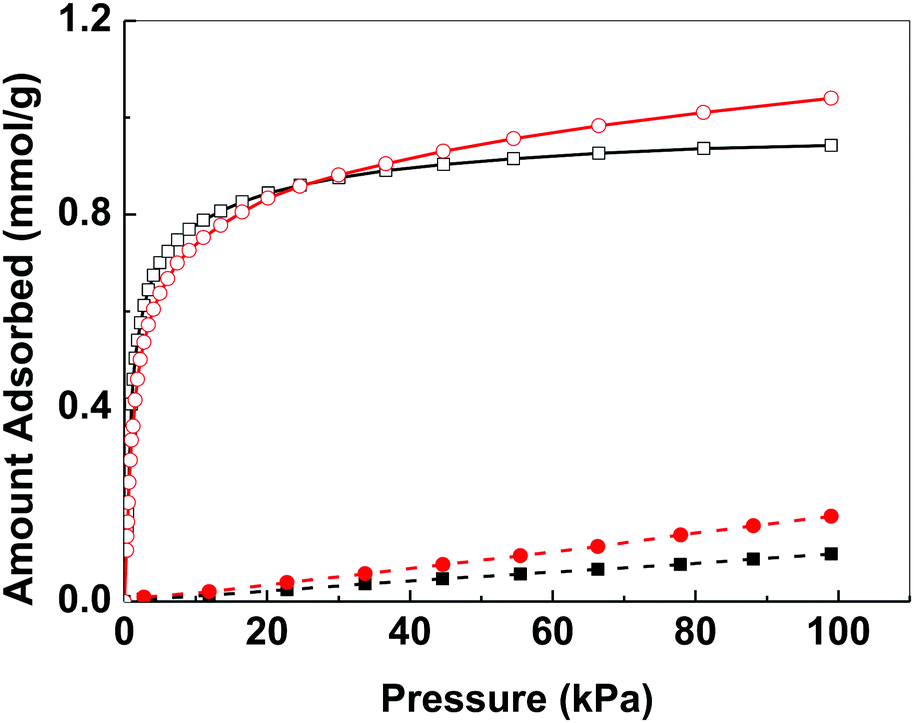 | ||
Fig. 4 Gas adsorption isotherms of bismuth subgallate recorded at 273 K (CO2: □, N2: ■) and 283 K (CO2:  , N2: , N2:  ). ). | ||
In conclusion, bismuth subgallate was revealed as a coordination polymer in contrast to the molecular formulas suggested in the past. The structure of bismuth subgallate significantly differs from those of other bismuth-based APIs such as bismuth subsalicylate,9d,e which forms isolated clusters, and bismuth subcitrate which can exist as an open framework.7a The material also displayed interesting gas sorption properties that warrant further investigations which may open up new application areas for bismuth compounds. Continuous rotation electron diffraction performed under low temperature was paramount for collecting data of sufficient quality. These results lead the way for the structure determination of other beam sensitive crystals, which is important for the development pharmaceutical compounds and understanding their functions.
This work was financially supported by the Swedish Research Council (VR), Swedish Governmental Agency for Innovation Systems (VINNOVA) through the Berzelii Center EXSELENT, MATsynCELL through the Röntgen-Ångström Cluster (VR), the Knut and Alice Wallenberg Foundation (KAW) for the project grant 3DEM-NATUR and a grant for purchasing the TEMs. A. K. I. is supported by KAW the through the MAX IV postdoctoral scholarship. We thank from Stockholm University Prof. Xiaodong Zou, Dr. Jekabs Grins, Dr. Stef Smeets and Dr. Zoltan Bacsik, and from Uppsala University Dr. Peng Zhang for discussions and assistance in data collection.
Notes and references
- C. Beck, J. Am. Med. Assoc., 1908, 50, 1233–1236 CrossRef.
- L.-M. Mai, C.-Y. Lin, C.-Y. Chen and Y.-C. Tsai, Biomaterials, 2003, 24, 3005–3012 CrossRef CAS PubMed.
- (a) S. L. Gorbach, Gastroenterol., 1990, 99, 863–875 CrossRef CAS; (b) M. Sparberg, Gastroenterol., 1974, 66, 476–480 CAS; (c) C.-Y. Lin, Y.-H. Shen, S.-H. Wu, C.-H. Lin, S.-M. Hwang and Y.-C. Tsai, Biochem. Biophys. Res. Commun., 2004, 315, 830–835 CrossRef CAS PubMed.
- (a) D. M. Keogan and D. M. Griffith, Molecules, 2014, 19, 15258–15297 CrossRef PubMed; (b) J. A. Salvador, S. A. Figueiredo, R. M. Pinto and S. M. Silvestre, Future Med. Chem., 2012, 4, 1495–1523 CrossRef CAS PubMed.
- (a) R. M. Peek and M. J. Blaser, Nat. Rev. Cancer, 2002, 2, 28–37 CrossRef CAS PubMed; (b) A. H. Mendis and B. J. Marshall, Biological Chemistry of Arsenic, Antimony and Bismuth, Wiley, John Wiley & Sons Ltd, Singapore, 2010, ch. 10 Search PubMed.
- J. M. Liou, M. S. Wu and J. T. Lin, J. Gastroenterol. Hepatol., 2016, 31, 1918–1926 CrossRef CAS PubMed.
- (a) W. Li, L. Jin, N. Zhu, X. Hou, F. Deng and H. Sun, J. Am. Chem. Soc., 2003, 125, 12408–12409 CrossRef CAS PubMed; (b) G. Tytgat, Digestion, 1987, 37, 31–41 CrossRef PubMed.
- (a) N. Chiba, Can. J. Gastroenterol. Hepatol., 2000, 14, 885–889 CAS; (b) D. Y. Graham and S.-Y. Lee, Gastroenterol. Clin. North Am., 2015, 44, 537–563 CrossRef PubMed.
- (a) Y. Yang, R. Ouyang, L. Xu, N. Guo, W. Li, K. Feng, L. Ouyang, Z. Yang, S. Zhou and Y. Miao, J. Coord. Chem., 2015, 68, 379–397 CrossRef CAS; (b) H. Sun, Biological chemistry of arsenic, antimony and bismuth, John Wiley & Sons Ltd, Chichester, UK, 2010 Search PubMed; (c) H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef CAS PubMed; (d) P. C. Andrews, G. B. Deacon, C. M. Forsyth, P. C. Junk, I. Kumar and M. Maguire, Angew. Chem., Int. Ed., 2006, 45, 5638–5642 CrossRef CAS PubMed; (e) V. André, A. Hardeman, I. Halasz, R. S. Stein, G. J. Jackson, D. G. Reid, M. J. Duer, C. Curfs, M. T. Duarte and T. Friščić, Angew. Chem., Int. Ed., 2011, 50, 7858–7861 CrossRef PubMed.
- N. Yang, Y. An, J. Cai, L. Hu, Y. Zeng, Z. Mao, G. Chen and H. Sun, Sci. China: Chem., 2010, 53, 2152–2158 CrossRef CAS.
- (a) U. Kolb, T. Gorelik, C. Kübel, M. Otten and D. Hubert, Ultramicroscopy, 2007, 107, 507–513 CrossRef CAS PubMed; (b) D. Zhang, P. Oleynikov, S. Hovmöller and X. Zou, Z. Kristallogr., 2010, 225, 94–102 CrossRef CAS; (c) W. Wan, J. Sun, J. Su, S. Hovmöller and X. Zou, J. Appl. Crystallogr., 2013, 46, 1863–1873 CAS.
- (a) Y. Yun, X. Zou, S. Hovmöller and W. Wan, IUCrJ, 2015, 2, 267–282 CrossRef CAS PubMed; (b) E. Van Genderen, M. Clabbers, P. Das, A. Stewart, I. Nederlof, K. Barentsen, Q. Portillo, N. Pannu, S. Nicolopoulos and T. Gruene, Acta Crystallogr., Sect. A: Found. Crystallogr., 2016, 72 Search PubMed; (c) M. D. Eddleston, K. E. Hejczyk, E. G. Bithell, G. M. Day and W. Jones, Chem. – Eur. J., 2013, 19, 7874–7882 CrossRef CAS PubMed.
- M. Gemmi, M. G. La Placa, A. S. Galanis, E. F. Rauch and S. Nicolopoulos, J. Appl. Crystallogr., 2015, 48, 718–727 CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- (a) L. Liu, L. N. Zakharov, J. A. Golen, A. L. Rheingold and T. A. Hanna, Inorg. Chem., 2008, 47, 11143–11153 CrossRef CAS PubMed; (b) D. Mendoza-Espinosa, A. L. Rheingold and T. A. Hanna, Dalton Trans., 2009, 5226–5238 RSC.
- D. Mansfeld, L. Miersch, T. Rüffer, D. Schaarschmidt, H. Lang, T. Böhle, R. W. Troff, C. A. Schalley, J. Müller and M. Mehring, Chem. – Eur. J., 2011, 17, 14805–14810 CrossRef CAS PubMed.
- (a) A. Ilyukhin and A. Poznyak, Crystallogr. Rep., 2000, 45, 56–63 CrossRef; (b) H. Wullens, B. Tinant, J.-P. Declercq and M. Devillers, Inorg. Chim. Acta, 2003, 343, 335–342 CrossRef CAS.
- (a) H. J. Breunig, N. Haddad, E. Lork, M. Mehring, C. Mügge, C. Nolde, C. I. Rat and M. Schürmann, Organometallics, 2009, 28, 1202–1211 CrossRef CAS; (b) P. C. Junk and L. M. Louis, Z. Anorg. Allg. Chem., 2000, 626, 556–559 CrossRef CAS; (c) L. J. Barbour, S. J. Belfield, P. C. Junk and M. K. Smith, Aust. J. Chem., 1998, 51, 337–342 CrossRef CAS; (d) A. Fridrichová, T. s. Svoboda, R. Jambor, Z. k. Padělková, A. Růžička, M. Erben, R. Jirásko and L. Dostál, Organometallics, 2009, 28, 5522–5528 CrossRef.
- Y. M. Yukhin, O. Logutenko, I. Vorsina and V. Evseenko, Theor. Found. Chem. Eng., 2010, 44, 749–754 CrossRef CAS.
- (a) M. Feyand, E. Mugnaioli, F. Vermoortele, B. Bueken, J. M. Dieterich, T. Reimer, U. Kolb, D. De Vos and N. Stock, Angew. Chem., Int. Ed., 2012, 51, 10373–10376 CrossRef CAS PubMed; (b) M. Savage, S. Yang, M. Suyetin, E. Bichoutskaia, W. Lewis, A. J. Blake, S. A. Barnett and M. Schröder, Chem. – Eur. J., 2014, 20, 8024–8029 CrossRef CAS PubMed; (c) A. Thirumurugan and A. K. Cheetham, Eur. J. Inorg. Chem., 2010, 3823–3828 CrossRef CAS; (d) A. K. Inge, M. Köppen, J. Su, M. Feyand, H. Xu, X. Zou, M. O'Keeffe and N. Stock, J. Am. Chem. Soc., 2016, 138, 1970–1976 CrossRef CAS PubMed.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
- A. Coelho, Bruker AXS, Karlsruhe, Germany, 2007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental procedures, elemental analysis, structure determination and refinement details, FT-IR spectrum, thermal analysis, XPD data, sorption isotherms, SEM images, electron diffraction images, Rietveld refinement plot, thermodiffraction, in situ XPD, acid stability and CIF. CCDC 1526756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc03180g |
| ‡ Crystallographic data: C7H9BiO8, Mr = 430.1, orthorhombic, Pmna, a = 8.5422(2), b = 4.66793(9), c = 23.7131(4) Å, V = 945.54(3) Å3, Z = 4, λ = 0.1.5406 Å (CuKα1), Rp = 11.8, Rwp = 16.43, Rexp = 9.41, GoF = 1.75. Structure determination with electron diffraction data and Rietveld refinement with X-ray powder diffraction data were conducted using SHELX24 and TOPAS25 respectively. CCDC 1526756 contains the supplementary crystallographic data for this paper.† |
| This journal is © The Royal Society of Chemistry 2017 |