
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA co-expression strategy to achieve labeling of individual subunits within a dimeric protein for single molecule analysis†
Fei
Lou
ab,
Jie
Yang
ab,
Si
Wu
*ab and
Sarah
Perrett
 *ab
*ab
aNational Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, 15 Datun Road, Chaoyang District, Beijing 100101, China. E-mail: sarah.perrett@cantab.net; wusi@moon.ibp.ac.cn
bUniversity of the Chinese Academy of Sciences, 19A Yuquan Road, Shijingshan District, Beijing 100049, China
First published on 19th June 2017
Abstract
A generic co-expression strategy for site-specific incorporation of a single donor–acceptor dye pair into any position in a dimeric protein, allowing single molecule FRET study of proteins previously inaccessible to this technique, such as the intrinsically disordered prion N-domain of Ure2 in the context of its globular dimeric C-domain.
Single molecule fluorescence resonance energy transfer (smFRET) has become a widely used tool to study the conformational heterogeneity and dynamics of biomacromolecules, which may be hidden and averaged in ensemble experiments.1,2 Furthermore, since smFRET enables measurement of the distances between pairs of labeling dyes, it can also provide structural information about biomolecules3,4 that may be hard to achieve using traditional structural biology methods such as crystallography; for example, the conformation of intrinsically disordered proteins (IDPs).5–8 IDPs are not only involved in many biological processes such as transcription, cell signal transduction and posttranslational modification,9 but are also associated with human diseases.10 The assembly of IDPs (i.e. proteins or peptides) into β-sheet rich amyloid aggregates is associated with human neurodegenerative diseases including Parkinson's and Alzheimer's. Single molecule FRET enables the direct investigation of the conformational flexibility and dynamics of IDPs, which contributes to understanding of their structure–function relationships. To observe individual biomolecules by smFRET, the sample needs to be labeled by suitable fluorophores at desired positions. The most widely used approach for labeling of a target protein is site-directed mutagenesis to introduce cysteine residues followed by labeling with maleimide-functionalized dyes.11 However, this method can only be applied to monomeric target proteins, while many proteins exist and execute functions in stable dimeric or oligomeric states,12 which makes it difficult to realize intramolecular smFRET measurements using classical cysteine labeling methods. In previous studies, in order to characterize the conformations and dynamics of dimeric or multimeric IDPs, the intrinsically disordered region (IDR) was usually truncated to obtain an isolated monomeric domain for smFRET experiments,13,14 which does not reflect the conformation of the original native state.
Here we present a strategy for labeling of dimeric proteins using the example of the dimeric protein Ure2. Ure2 is the protein determinant of the Saccharomyces cerevisiae prion state [URE3]15 and is an important model for study of prion propagation and amyloid formation. It is a homodimeric protein in its native state and each chain consists of two domains. The N-terminal domain (residue 1–93) of Ure2 is an intrinsically disordered region which plays a key role in the prion phenomenon in vivo and is responsible for the ability to assemble into amyloid fibrils in vitro.16 The C-terminal globular domain (residue 94–354), which contains the dimerization interface, shows high structural similarity to glutathione transferase enzymes17 and participates in negative regulation of nitrogen metabolism.18 The existence of interactions between the N-terminal prion domain and C-terminal globular domain has long been under debate, with both positive19–22 and negative23,24 indications obtained from previous studies. The observation that the level of enzyme activity of the C-terminal domain is influenced by the presence of the N-terminal prion domain25 and the finding that fibril formation kinetics of the isolated N-terminal is greatly accelerated compared to that of full-length Ure2,26 suggest that the two domains may influence one another. Direct characterization of the conformation and dynamics of the N-terminal domain of Ure2 would facilitate understanding of its relationship with the adjacent C-terminal domain and its conversion to a highly ordered amyloid state. However, since the disordered N-terminal domain is highly flexible and aggregation-prone, the structure of full-length native Ure2 is not readily accessible by high resolution structural biology techniques such as NMR and X-ray crystallography. Thus smFRET is the most suitable approach for characterization of the IDR of Ure2.
Since native Ure2 is a dimer, we faced the challenge mentioned above that it is difficult to introduce the mutation or labeling site into just one chain of the dimer by conventional mutagenesis. Subunit exchange between WT homomultimers and Cys-mutated homomultimers has often been used as a strategy to obtain heteromultimers in previous smFRET studies.13,27 However, because the Ure2 dimer is extremely stable and does not readily dissociate,28 sufficient quantity of heterodimers could not be obtained for further smFRET experiments (Fig. S1, ESI†). In order to solve this problem, we applied a new strategy that could readily produce heterodimers during protein expression, with the pair of labeling sites either in the same monomer or in different monomers within the dimer, making it possible to measure the distances between any residue pair within the dimeric protein. We took advantage of the pQLink co-expression system which is used to form multi-component protein complexes by co-expression of the subunits,29 to simultaneously express both His-tagged WT Ure2 and the untagged double-Cys Ure2 mutant, resulting in production of three types of dimers (Fig. 1A). After purification by nickel column, the untagged double-Cys/double-Cys homodimer could be removed, leaving the WT/WT homodimer and the target WT/double-Cys heterodimer. As the WT/WT homodimer (which lacks any Cys residues) cannot be labeled by maleimide functionalized dyes, its presence is silent to FRET analysis. Thus, the single pair FRET labeling only occurs in the WT/double-Cys heterodimer, which allows us to study the relative conformation of different regions of a single monomer within the dimer. To study FRET pairs located in different subunits, and thus to obtain a heterodimer with different Cys mutations in each polypeptide chain, we added a His-tag to one mutant polypeptide chain and a Strep-tag to the other mutant chain, and then carried out a Strep-tag affinity purification step subsequent to nickel column purification (Fig. 1B).
 | ||
| Fig. 1 Scheme of the co-expression strategy to obtain the heterodimers of Ure2. (A and B) The vector maps of the pQLink co-expression systems (upper panels) and purification protocols (lower panels) for production of heterodimers of Ure2 with two mutation sites in one chain (A) and with a mutation site in each chain (B). Green, prion domain of Ure2; yellow, C-terminal domain of Ure2; red star, the Cys mutation sites in Ure2. The numbers shown indicate the theoretical ratio of the different dimers at each stage of protein expression and purification. | ||
In order to validate the co-expression labeling strategy, we first inserted WT-Ure2 and the Ure2 mutant V9C/F295C into the pQLink co-expression vector, in order to produce the heterodimer. We observed two bands by SDS-PAGE (Fig. 2A), of which the upper band represents His-tagged WT Ure2 and the lower band represents the untagged Ure2 mutant V9C/F295C. Q-TOF mass spectrometry (MS) was performed to confirm the molecular weight of the heterodimer (Fig. 2B). The chromatography and ionization process of MS causes dimer dissociation, and so the monomer peaks are observed. Q-TOF MS indicates that the molecular weight of His-tagged WT Ure2 is 41774 Da and the untagged V9C/F295C mutant is 40375 Da, in agreement with the theoretical MW values. The intensity ratio of the two peaks is about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, which is consistent with the expected expression ratio of WT/WT, WT/Cys and Cys/Cys dimers of 1:2:1, of which only WT/WT and WT/Cys dimers contain a His-tag and so are present in the purified mixture subjected to MS. These results indicate that the heterodimer forms during expression and could be purified through a single His-tag.
:2, which is consistent with the expected expression ratio of WT/WT, WT/Cys and Cys/Cys dimers of 1:2:1, of which only WT/WT and WT/Cys dimers contain a His-tag and so are present in the purified mixture subjected to MS. These results indicate that the heterodimer forms during expression and could be purified through a single His-tag.
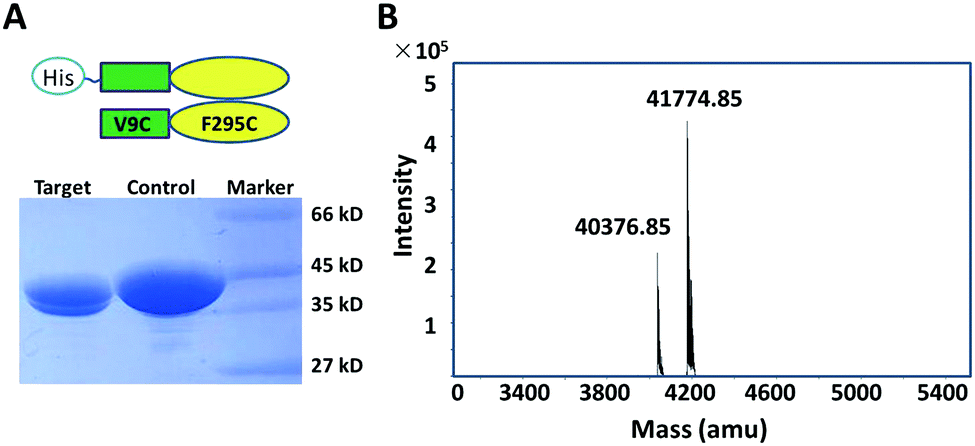 | ||
| Fig. 2 Characterization of Ure2 heterodimers by SDS-PAGE and Q-TOF. (A) SDS-PAGE of purified Ure2 mutants. Lane 1, Ure2 WT/V9C-F295C heterodimer; lane 2, homodimeric His-tagged WT-Ure2. (B) Q-TOF mass spectrum of the heterodimer of WT/V9C-F295C. amu, atomic mass units. | ||
To further check whether the protein purified by our strategy contains any unwanted Cys/Cys homodimer, we obtained a single Cys mutant WT/V9C heterodimer using the above strategy using the pQLink vector, and obtained V9C/V9C homodimer (using a standard pRSET vector) as a positive control. We labeled the above samples with a mixture of Alexa Fluor 555 (AF555) and Alexa Fluor 647 (AF647) and carried out smFRET measurements. In this situation, only the V9C/V9C homodimer will give a FRET signal. As shown in Fig. 3, the WT/V9C heterodimer does not show a significant FRET distribution (Fig. 3A), while the V9C/V9C dimer gives a FRET peak centred at 0.59, reflecting the distance between the labeled V9C residues in each monomer within the dimer (Fig. 3B). Therefore, the above results confirm that we can obtain the heterodimer of Ure2 without contamination of Cys/Cys homodimer by the co-expression strategy.
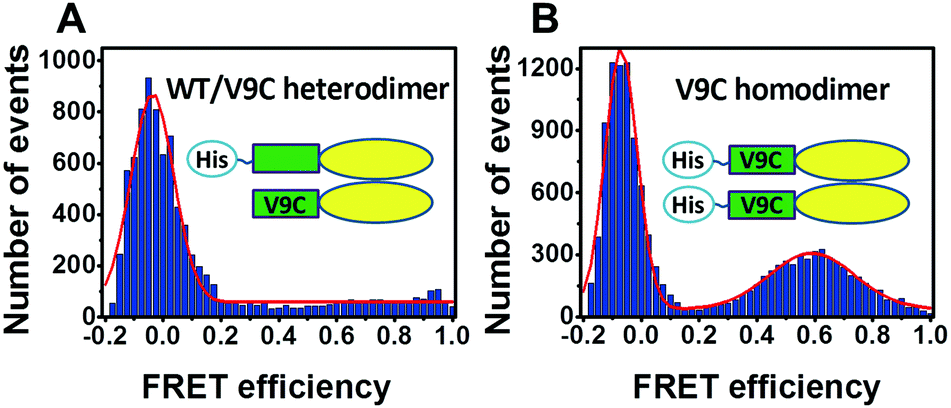 | ||
| Fig. 3 Comparison of FRET signal of hetero- and homo-dimeric Ure2. Single-molecule FRET histograms of Ure2 WT/V9C heterodimer (A) and Ure2-V9C homodimer (B). The histograms are fitted to either single (A) or double (B) Gaussian functions (red lines). | ||
Using the above strategy, we constructed multiple labeling variants for smFRET study. We chose Val9, Ser26, and Ser52 in the N-terminal domain in combination with Glu108, Lys240, and Phe295 in the C-terminal domain as the Cys mutations and labeling sites to construct a total of nine variants each with a labeling dye pair within the same monomer. We also constructed V9C/L351C, S26C/L351C, and S52C/F295C, where the two Cys mutations are in different chains of the dimer (Fig. 4A and Table S1, ESI†). The selected C-terminal residues are broadly distributed within the structure, in order to allow unambiguous triangulation of N-terminal residues. We then applied smFRET to measure the intramolecular distances between the fluorescent dye pairs. By triangulation, the spatial positions of fluorophore labels on the disordered N-terminal prion domain could be determined based on the already known structure of the C-terminal domain. The smFRET results show that all of the three selected N-terminal residues have higher FRET efficiency to Lys240 than to other labeled C-terminal residues (Fig. 4B and Table S1, ESI†). It should be noted that the population with FRET efficiency near zero is a result of the presence of molecules without an active acceptor, and so is not included in the analysis. With the assistance of nanopositioning system tools30 based on the combined smFRET data (Table S1, ESI†) and fluorescence anisotropy data (Table S2, ESI†), we determined the probability density distributions of the fluorescent dyes attached to the three N-terminal residues (Val9, Ser26 and Ser52) (Fig. 5). The spatial profile of the three positions is located near the upper-side of the Ure2 C-terminal domain but does not wrap around or extend far away from the C-terminal domain. Moreover, the probability density maps of the three labeled N-terminal residues partially overlap, indicating the prion domain does not adopt an extended conformation. We also measured the smFRET of the V9C/S52C mutant which showed a high FRET efficiency of about 0.8 (Fig. S2, ESI†), in agreement with the above smFRET result that the N-terminal domain is not extended.
 | ||
| Fig. 4 Single-molecule FRET analysis of Ure2 heterodimer variants. (A) Mutation and labeling sites in C-terminal domain of Ure2 (PDB [1G6W]), including Glu108, Arg240, Phe295 and Leu351 residues shown as spheres. (B) smFRET histograms of twelve variants as labeled in the figure. The histograms are fitted to a double Gaussian function. | ||
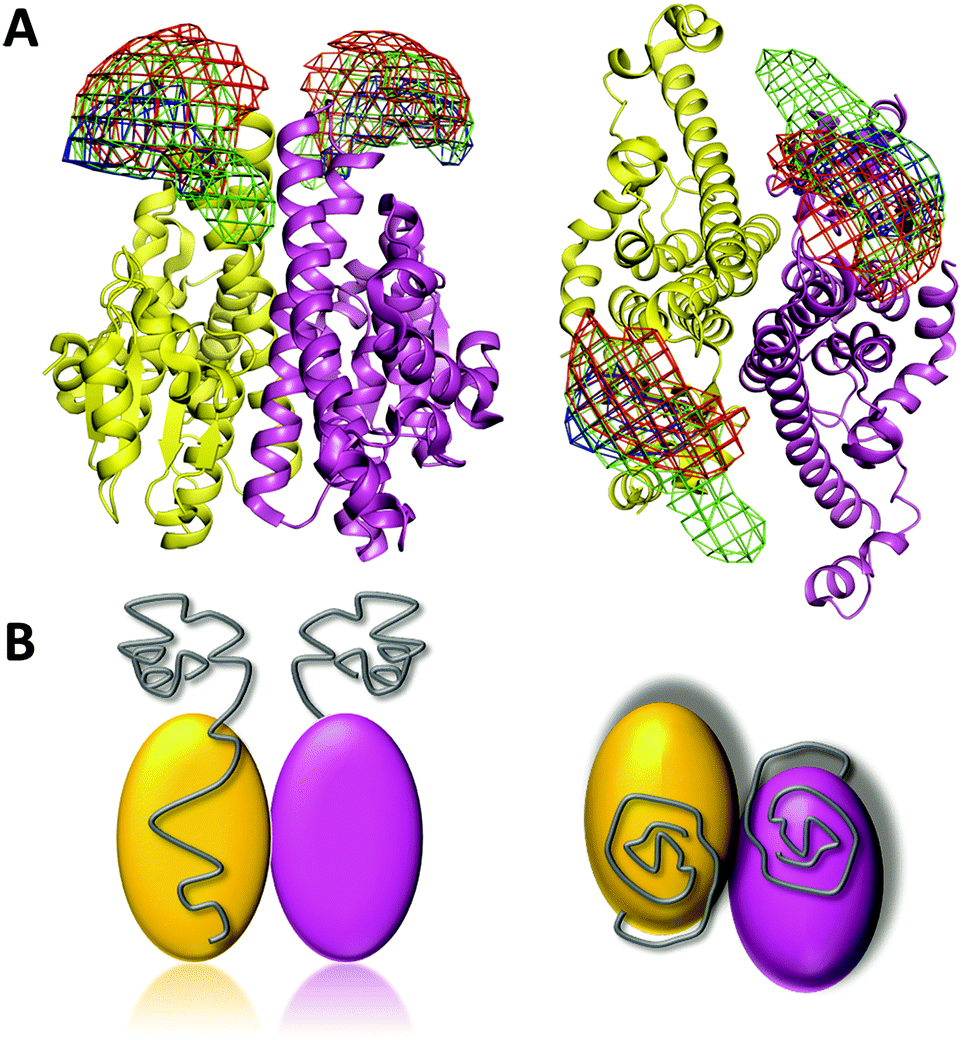 | ||
| Fig. 5 Spatial distribution of the Ure2 prion domain. (A) The density map shows the spatial distributions of Val9 (red), Ser26 (green) and Ser52 (blue) residues located in the N-terminal prion domain, in relation to the globular C-terminal domain (PDB [1G6W]). Left panel, side view; right panel, top view. (B) Cartoon model of the full-length Ure2 protein with the location of the N-terminal prion domains shown in grey. Left panel, side view; right panel, top view. | ||
To identify and analyse the conformational dynamics of the prion domain, we performed fluorescence correlation spectroscopy (FCS) experiments with Cy3B/AF647 dual-labeled variants, and also Cy3B-only labeled variants as a control. The results show that the FRET fluctuation between the prion domain and the C-terminal domain is less than 7% (Fig. S3, ESI†), suggesting that there is no significant fast dynamics of the prion domain and the broadening of the smFRET histogram can be attributed to conformational heterogeneity or slow fluctuation. This result is in contrast to the monomeric IDP α-synuclein6 and the truncated IDR of the yeast prion protein Sup3514 which both show significant fast dynamics of the disordered chain. The lack of fast dynamics of the N-terminal domain of Ure2 suggests that the presence of the C-terminal globular domain can to some extent stabilize the N-terminal disordered prion domain by weak interactions. We then labeled Ure2 K240C with a photoactive crosslinker benzophenone-4-maleimide (BPM), as the smFRET results indicate that the prion domain is close to this residue, and detected the crosslinking product between K240 and the Ure2 N-terminal domain by MALDI-TOF mass spectrometry after UV irradiation. An N-terminal fragment, residues 2–17, was detected to be crosslinked to K240C-BPM (Fig. S4, ESI†), which further supports the spatial location of the prion domain and possible interdomain interactions indicated by the smFRET results.
In conclusion, we have demonstrated a co-expression strategy to obtain dimers of Ure2 containing monomer-specific labels in order to study the intramolecular conformation by smFRET. By this method, we found that the prion domain of Ure2 adopts an ensemble of conformations and lacks fast dynamics. The probable spatial position of the prion domain of Ure2 is also presented, although a more precise description of the structure and conformation will require further investigation in combination with other techniques, for example more extensive cross-linking and molecular dynamics simulations. As a proof-of-principle study, our strategy allows the measurement of distances between any residue pair within an individual monomer or between two monomers within a dimer, which cannot readily be achieved by conventional Cys mutagenesis and labeling methods. This provides a new approach to probe the structure or conformation of IDPs that are not monomeric, and also has potential applications in the smFRET study of other dimeric proteins that have important biological functions, such as GPCRs.
We thank Prof. Cai Zhang (IBP, CAS) for providing plasmids, Dr Xun Li (Nikon Instruments Co. Ltd) for helpful discussions, Prof. Jens Michealis (University of Ulm) for advice on FRETnpsTools, and Zhensheng Xie and Lili Niu (IBP, CAS) for assistance with MS experiments. We acknowledge support from the MoST 973 Program [2013CB910700], the NSFC [21673278, 31110103914, 31300631] and the CEBioM.
References
- A. Mashaghi, G. Kramer, D. C. Lamb, M. P. Mayer and S. J. Tans, Chem. Rev., 2014, 114, 660–676 CrossRef CAS PubMed.
- B. Schuler, J. Nanobiotechnol., 2013, 11(Suppl 1), S2 CrossRef PubMed.
- S. Kalinin, T. Peulen, S. Sindbert, P. J. Rothwell, S. Berger, T. Restle, R. S. Goody, H. Gohlke and C. A. Seidel, Nat. Methods, 2012, 9, 1218–1225 CrossRef CAS PubMed.
- A. T. Brunger, P. Strop, M. Vrljic, S. Chu and K. R. Weninger, J. Struct. Biol., 2011, 173, 497–505 CrossRef CAS PubMed.
- S. Elbaum-Garfinkle and E. Rhoades, J. Am. Chem. Soc., 2012, 134, 16607–16613 CrossRef CAS PubMed.
- A. C. Ferreon, Y. Gambin, E. A. Lemke and A. A. Deniz, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5645–5650 CrossRef CAS PubMed.
- L. A. Metskas and E. Rhoades, J. Am. Chem. Soc., 2015, 137, 11962–11969 CrossRef CAS PubMed.
- M. Brucale, B. Schuler and B. Samori, Chem. Rev., 2014, 114, 3281–3317 CrossRef CAS PubMed.
- M. M. Babu, Biochem. Soc. Trans., 2016, 44, 1185–1200 CrossRef CAS PubMed.
- M. M. Babu, R. van der Lee, N. S. de Groot and J. Gsponer, Curr. Opin. Struct. Biol., 2011, 21, 432–440 CrossRef CAS PubMed.
- K. L. Holmes and L. M. Lantz, Methods Cell Biol., 2001, 63, 185–204 CrossRef CAS PubMed.
- N. J. Marianayagam, M. Sunde and J. M. Matthews, Trends Biochem. Sci., 2004, 29, 618–625 CrossRef CAS PubMed.
- F. Huang, S. Rajagopalan, G. Settanni, R. J. Marsh, D. A. Armoogum, N. Nicolaou, A. J. Bain, E. Lerner, E. Haas, L. Ying and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20758–20763 CrossRef CAS PubMed.
- S. Mukhopadhyay, R. Krishnan, E. A. Lemke, S. Lindquist and A. A. Deniz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2649–2654 CrossRef CAS PubMed.
- R. B. Wickner, Science, 1994, 264, 566–569 CAS.
- K. L. Taylor, N. Cheng, R. W. Williams, A. C. Steven and R. B. Wickner, Science, 1999, 283, 1339–1343 CrossRef CAS PubMed.
- L. Bousset, H. Belrhali, J. Janin, R. Melki and S. Morera, Structure, 2001, 9, 39–46 CrossRef CAS PubMed.
- D. Blinder, P. W. Coschigano and B. Magasanik, J. Bacteriol., 1996, 178, 4734–4736 CrossRef CAS PubMed.
- D. C. Masison, M. L. Maddelein and R. B. Wickner, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12503–12508 CrossRef CAS.
- E. Fernandez-Bellot, E. Guillemet, A. Baudin-Baillieu, S. Gaumer, A. A. Komar and C. Cullin, Biochem. J., 1999, 338(Pt 2), 403–407 CrossRef CAS PubMed.
- M. L. Maddelein and R. B. Wickner, Mol. Cell. Biol., 1999, 19, 4516–4524 CrossRef CAS PubMed.
- E. Fernandez-Bellot, E. Guillemet and C. Cullin, EMBO J., 2000, 19, 3215–3222 CrossRef CAS PubMed.
- M. M. Pierce, U. Baxa, A. C. Steven, A. Bax and R. B. Wickner, Biochemistry, 2005, 44, 321–328 CrossRef CAS PubMed.
- C. Thual, L. Bousset, A. A. Komar, S. Walter, J. Buchner, C. Cullin and R. Melki, Biochemistry, 2001, 40, 1764–1773 CrossRef CAS PubMed.
- Z. R. Zhang and S. Perrett, J. Biol. Chem., 2009, 284, 14058–14067 CrossRef CAS PubMed.
- A. Loquet, L. Bousset, C. Gardiennet, Y. Sourigues, C. Wasmer, B. Habenstein, A. Schutz, B. H. Meier, R. Melki and A. Bockmann, J. Mol. Biol., 2009, 394, 108–118 CrossRef CAS PubMed.
- C. Ratzke, M. Mickler, B. Hellenkamp, J. Buchner and T. Hugel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16101–16106 CrossRef CAS PubMed.
- D. Galani, A. R. Fersht and S. Perrett, J. Mol. Biol., 2002, 315, 213–227 CrossRef CAS PubMed.
- C. Scheich, D. Kummel, D. Soumailakakis, U. Heinemann and K. Bussow, Nucleic Acids Res., 2007, 35, e43 CrossRef PubMed.
- A. Muschielok, J. Andrecka, A. Jawhari, F. Bruckner, P. Cramer and J. Michaelis, Nat. Methods, 2008, 5, 965–971 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc03032k |
| This journal is © The Royal Society of Chemistry 2017 |