
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Turning on the red phosphorescence of a [Ru(tpy)(bpy)(Cl)]Cl complex by amide substitution: self-aggregation, toxicity, and cellular localization of an emissive ruthenium-based amphiphile†
B.
Siewert
 ab,
M.
Langerman
a,
Y.
Hontani
c,
J. T. M.
Kennis
c,
V. H. S.
van Rixel
a,
B.
Limburg
a,
M. A.
Siegler
d,
V.
Talens Saez
a,
R. E.
Kieltyka
a and
S.
Bonnet
*a
ab,
M.
Langerman
a,
Y.
Hontani
c,
J. T. M.
Kennis
c,
V. H. S.
van Rixel
a,
B.
Limburg
a,
M. A.
Siegler
d,
V.
Talens Saez
a,
R. E.
Kieltyka
a and
S.
Bonnet
*a
aLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 233CC Leiden, The Netherlands. E-mail: bonnet@chem.leidenuniv.nl
bInstitute of Pharmacy, Pharmacognosy, University of Innsbruck, CCB, Innrain 80-82, 6020 Innsbruck, Austria
cDepartment of Physics and Astronomy, Vrije Universiteit, De Boelelaan 1081, 1081HV Amsterdam, The Netherlands
dSmall Molecule X-ray Facility, Department of Chemistry Johns Hopkins University, Baltimore, MD 21218, USA
First published on 28th June 2017
Abstract
Coupling the notoriously non-emissive complex [Ru(tpy)(bpy)Cl]Cl (tpy = 2,2′:6′,2′′-terpyridine, bpy = 2,2′-bipyridine) to a C12 alkyl chain via an amide linker on the 4′ position of the terpyridine yielded a new amphiphilic ruthenium complex showing red emission and chloride-dependent aggregation properties. This emissive complex is highly cytotoxic in A549 non-small lung cancer cells where it can be followed by confocal microscopy. Uptake occurs within minutes, first by insertion into the cellular membrane, and then by migration to the peri-nuclear region.
Over 80 years ago Francis Burstall reported a reaction between RuCl3 and 2,2′ bipyridine at 250 °C yielding the red salt [Ru(bpy)3]Cl2 ([1]Cl2).1 The stability of this complex, coupled to its unique electrochemical and photophysical properties, has led to the development of a wide class of octahedral ruthenium polypyridyl complexes2 and countless reports on their catalytic,2c,3 photosensitizing,4 or biological applications.5 Depending on the energy level of their triplet excited states, these complexes may be phosphorescent or not. Generally, complexes with three 2,2′-bipyridine (bpy) ligands, e.g. [Ru(bpy)3]Cl2 ([1]Cl2), show a strong emission,2a while complexes containing tridendate 2,2′;6′,2′′-terpyridine ligands (tpy), e.g. [Ru(tpy)2]Cl2 ([2]Cl2), show no emission at room temperature. Negligible phosphorescence was observed for complexes containing both a tpy and a bpy ligand, e.g. [Ru(tpy)(bpy)Cl]Cl ([3]Cl).2a,c,6 Emission in [1]2+ is due to the high energy gap between the triplet metal-centred (3MC) and triplet metal-to-ligand charge transfer (3MLCT) excited states (+42.7 kJ mol−1).7 In tpy-containing complexes [2]2+ and [3]+ this 3MC-3MLCT energy gap decreases to −16.7 kJ mol−1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and −6.6 kJ mol−1, respectively,9 which promotes non-radiative decay via the 3MC states (Fig. 1). Thermal promotion of the 3MC state from the photochemically generated 3MLCT state may also lead to ligand photosubstitution, in particular for [Ru(tpy)(bpy)(L)]2+ complexes (L is a monodentate ligand),5e,10 which generates in water the aqua complex [Ru(tpy)(bpy)(OH2)]2+ ([6]2+).
8 and −6.6 kJ mol−1, respectively,9 which promotes non-radiative decay via the 3MC states (Fig. 1). Thermal promotion of the 3MC state from the photochemically generated 3MLCT state may also lead to ligand photosubstitution, in particular for [Ru(tpy)(bpy)(L)]2+ complexes (L is a monodentate ligand),5e,10 which generates in water the aqua complex [Ru(tpy)(bpy)(OH2)]2+ ([6]2+).
 | ||
| Fig. 1 Top: Chemical structures of [Ru(bpy)3]Cl2 ([1]Cl2), [Ru(tpy)2]Cl2 ([2]Cl2), and [Ru(tpy)(bpy)Cl]Cl ([3]Cl). Bottom: Simplified Jablonski diagram of RuII polypyridine complexes. The ΔE(3MC–3MLCT) decreases from [1]2+ > [2]2+ ≈ [Ru(tpy)(bpy)L]2+, favouring non-radiative decay for terpyridine-containing complexes via thermally driven population of the 3MC state from the photochemically generated 3MLCT state. (GS = ground state, MLCT = metal-to-ligand charge transfer, isc = intersystem crossing, MC = metal-centred.) | ||
When generated in a cell, aqua complexes such as [6]2+ may react with biological nucleophiles such as amino acids, peptides (e.g. glutathione), protein residues, or DNA base pairs,11 which – as studied for cisplatin – may lead to cell death. Thus, light-sensitive complexes of the [Ru(tpy)(bpy)(L)]2+ type represent an interesting class of pro-drugs: triggering the formation of the active species [6]2+ by an external, human-controlled process (i.e. light irradiation), allows for selective drug activation in illuminated tumour tissue, a concept known as photo-activated anticancer chemotherapy (PACT).12,13 However, as discussed by Alessio,14 low cellular uptake is an obstacle for charged ruthenium complexes. If the pro-drug does not reach the nucleus, how could lethal metal–DNA adducts be formed? We recently reported that increasing the lipophilicity of a [Ru(tpy)(bpy)(L)]2+ complex by using a cholesterol–thioether ligand L led to a significant increase in cellular uptake, self-aggregating properties, and dark cytotoxicity.12a However, a long pre-irradiation incubation time of 24 h was necessary to observe any phototoxicity, and prodrug localization was impossible to probe in vitro because of the non-luminescent character of the ruthenium compound. Here, inspired by Heinze et al.15 we increased the lipophilicity of [3]Cl by conjugating a C12 alkyl tail in 4′ position of the tpy ligand via an amide linker, and studied whether such mildly electron-withdrawing groups would lead to increased phosphorescence. This communication reports on the synthesis of the amphiphilic complex [Ru(Rtpy)(bpy)Cl]Cl ([4]Cl, Fig. 2, Rtpy = N-dodecyl-[2,2′:6′,2′′-terpyridine]-4′-carboxamide), its aggregation properties in aqueous solution, and its significantly improved phosphorescence, which allows for following uptake and localization in vitro.
 | ||
| Fig. 2 Top: Chemical formula of complex [4]Cl, and proton numbering scheme for 1H NMR attribution. Bottom: Displacement ellipsoid plot (50% probability level) of cationic [4]+ as observed in the crystal structure of [4]Cl·H2O·C3H6O at 110(2) K. The counter-anion and lattice solvent molecules have been omitted for clarity. Characteristic bond lengths [Å]: Ru1–N1 = 2.077(3), Ru1–N2 = 2.049(3), Ru1–N3 = 2.066(3), Ru1–N4 = 1.954(3), Ru1–N5 = 2.076(3), Ru1–Cl = 2.3956(8). | ||
Complex [4]Cl was synthesized in three steps by amide coupling of dodecylamine and 4′-carboxy-2,2′;6′,2′′-terpyridine, followed by coordination to ruthenium trichloride and 2,2′-bipyridine coordination in reductive conditions (Fig. S1, ESI†). 1H NMR experiments disclosed the typical downfield shift of the bpy proton A6, as well as the upfield shift of the B6 proton due to the shielding cone of the tpy ligand. Furthermore, the 3′ proton on the amide-functionalized tpy appeared downfield-shifted (9.21 ppm in d6-DMSO, Fig. S3, ESI†) compared to the non-substituted complex [3]+ (8.81 ppm, d6-DMSO). The doubled triplet at 3.46 ppm (3J = 6.7 Hz, 3J = 6.5 Hz) was assigned to the alpha methylene group of the aliphatic chain due to its coupling with the amide proton. A crystal structure of [4]Cl·H2O·C3H6O was obtained revealing, next to the typical octahedral environment of the ruthenium complexes, a trans-conformation for the amide bond (Fig. 2 and Fig. S4, ESI†), and hydrogen bonding between the chloride counter anion, the water solvate molecule, and the amide group. Finally elemental analysis confirmed the chemical purity of the bulk compound.
In spite of its long alkyl tail [4]Cl was found soluble in aqueous solutions. In water, its UV-vis spectrum (Fig. 3) showed a broad 1MLCT transition (dπ → π*, see HOMO and LUMO in Fig. S20, ESI†) with an absorbance maximum bathochromically shifted (502 nm) compared to [3]Cl (484 nm).16
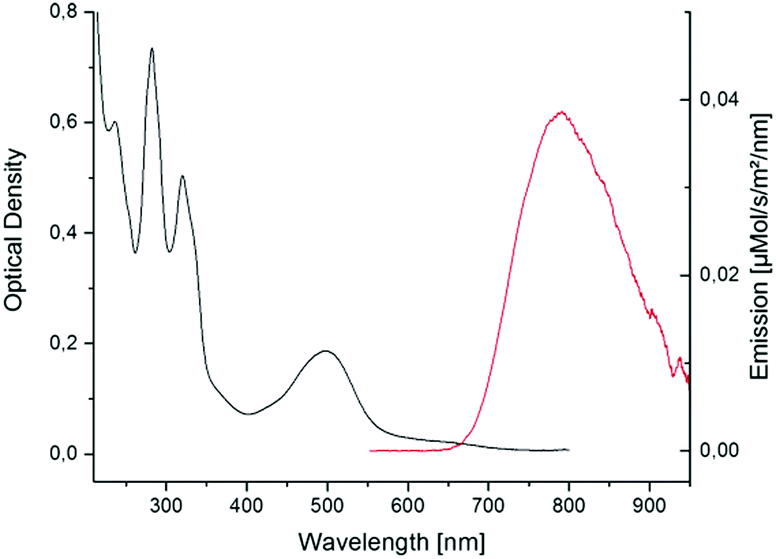 | ||
| Fig. 3 Absorption and emission spectra of [4]+ in MilliQ water and under air. Excitation wavelength was 455 nm (power 50 mW). | ||
Remarkably, [4]Cl was also emissive (Fig. 3). In MilliQ water and under air its emission spectrum showed a maximum at 791 nm with an emission quantum yield in air of ϕphos = 0.0004. This quantum yield is two orders of magnitude lower as compared to the reference compound [1]Cl2 (λmax,em = 629 nm, ϕphos,ref = 0.04),17 but it is still much higher than for [3]Cl, for which quantum yield measurements were impossible.2a The emission intensity increased proportionally to the concentration until 60 μM, indicating no self-quenching in this concentration range (Fig. S7, ESI†).18 Furthermore, the emission spectra in acetonitrile, methanol, or pentane, proved that the emission of [4]Cl in water was not caused by aggregation. The observation of red-shifted emission for [4]Cl compared to [3]Cl indicates that the 3MLCT band lies at significantly lower energy than the 3MC band, which also explains the increased phosphorescence quantum yield: the 3MC–3MLCT gap is higher in [4]+ than in [3]+, leading to less phosphorescence quenching. Transient absorption spectroscopy (TAS) and time-correlated single photon counting (TCSPC) experiments were done in PBS under air at 7.5 μM and 75 μM, respectively, to investigate the excited state lifetimes of [4]Cl. In PBS, the high chloride concentration prevented formation of the aqua complex [Ru(Rtpy)(bpy)(OH2)]2+ ([7]2+, see Fig. S9, ESI†) that, when formed in pure water, was also emissive (Fig. S8, ESI†). Global analysis of the TCSPC data (see Fig. S11 and Table S4, ESI†) indicated that the decay of emission of [4]Cl in PBS can be best described by three lifetimes, i.e. τ3 = 0.44 ns (44%), τ4a = 9 ns (14%), and τ4b = 40 ns (42%). TAS (see Fig. S10, ESI†), which investigated photon absorption, revealed next to two emission-relevant lifetimes (i.e. τ3 = 110 ps, τ4 = 17 ns) three lifetimes of non-emissive components (i.e. τ1 < 50 fs, τ2 ∼ 3 ps, and τ5 = 200 ns). The ultrafast lifetime (i.e. τ1 < 50 fs) might correspond to the intersystem crossing 1MLCT → 3MLCT, which is known to lie in the fs range for ruthenium polypyridyl complexes.19 The second lifetime (τ2 ∼ 3 ps) was assigned to the vibrational cooling of the 3MLCT(tpy) state, accordingly to reported values.10c The longest component (τ5 = 200 ns) can be assigned to the non-radiative decay from the excited 3MC states to the GS. The shorter of the two emissive components, i.e. τ3 = 110 ps, showed a slight blue shift and can be correlated to the 0.44 ns component found in TCSPC. Most likely, this lifetime belongs to interconversion (IC) between the 3MC and 3MLCT excited states. The main decay found in TAS, i.e. τ4 = 17 ns, equals the radiative decay from the 3MLCT excited state to the ground state, and was more resolved in TCSPC, where it split into a major (i.e. τ4b = 40 ns (42%)) and a minor (i.e. τ4a = 9 ns (14%)) component, reflecting the several possible emissive 3MLCT states of ruthenium polypyridyl complexes.20 A schematic overview of the excited state interconversion for compound [4]Cl in PBS is given in Fig. S12, ESI.†
In order to investigate the non-radiative decay originating from the 3MC excited states, green light irradiation (490 nm) in water was followed by UV-vis spectroscopy (Fig. S13, ESI†). The kinetics of chloride substitution in [4]+ by water, to form the aqua species [Ru(Rtpy)(bpy)(OH2)]2+ ([7]2+), was compared with dark conditions. In the dark, thermal hydrolysis was slow, characterized by a hydrolysis rate constant of kdarkhyrdo = 7 × 10−5 s−1. Under green light irradiation the rate constant was enhanced by a factor 24 to k490nmhyrdo = 2 × 10−3 s−1, corresponding to a photosubstitution quantum yield of 0.0176 (see ESI† part 2.4, Fig. S14). The acceleration of hydrolysis under light irradiation confirmed the TAS results, and indicated that for [4]+ the 3MC excited states are thermally accessible, at room temperature, from the photogenerated 3MLCT states.
Further, it was anticipated that the attachment of a long alkyl tail to the 4′-position on the tpy ligand of [4]+ may lead to self-assembly.31 The morphology of a self-assembled structure can be predicted by calculating the packing parameter P of the surfactant defined as ν/(l·a), where ν is the volume (in Å3) and l the length (in Å) of the aliphatic chain, while a represents the area of the head group (in Å2).21 Employing this equation with values calculated as described in Section 3 of the ESI,† the packing parameter P was 0.2. Therewith the assembly of [4]+ as spherical micelles is predicted,22 which was studied by DLS and microscopy. Self-assembly of [4]Cl in aqueous solutions was found to highly depend on chloride concentration. Dynamic light scattering (DLS) measurements of [4]Cl in PBS (200 μM) displayed a highly disperse collection of aggregate sizes ranging from a few nanometers to microns in hydrodynamic diameter (dH, see Fig. S16, ESI†). These observations were corroborated by confocal microscopy at a lower concentration (5 μM), which showed the formation of micron-sized, phosphorescent aggregates of micelles (Fig. 4). Zeta potential measurements suggested the formation of charged assemblies (ζ = +19.03 ± 11.1 mV). In contrast, dissolution of [4]Cl (200 μM) in MilliQ water showed only a single population of stable micelles with a hydrodynamic radius dH of 45 nm, as measured by DLS and further supported by cryo-TEM (Fig. S17, ESI†). Zeta potential measurements showed a 37% increase in charge to ζ = +26.1 ± 7.3 mV. This increased ζ value may be rationalized by the hydrolysis of the Ru-Cl bond of [4]+ in pure water to form the bicationic aqua complex [7]2+, increasing stability of the micellar structure, however the effects of buffer salts on the measured values cannot be excluded (Fig. S16, ESI†). Moreover, the lack of secondary aggregation in pure water as opposed to PBS where micron-sized phosphorescent assemblies were observed (Fig. 4), suggested that micelle aggregation in PBS is driven by the high chloride (137 mM) concentration that stabilizes coordination of the chloride ligand to ruthenium. Similar aggregation phenomena have been documented for other metallo-surfactants.23 In addition, amide bridging by the chloride counter-ions may affect the secondary aggregation process. Overall, these studies reveal that the self-assembly of [4]Cl in aqueous solutions can be modulated by the chloride concentration.
 | ||
| Fig. 4 Left: Confocal fluorescent micrograph of [4]Cl in PBS (5 μM) in absence of fetal bovine serum (FBS) showing micellar aggregation. Right: Confocal fluorescent micrograph of A549 cells incubated with [4]Cl (5 μM) for 15 minutes. | ||
Our group recently demonstrated that non-emissive ruthenium amphiphiles can be highly toxic to cancer cells by destabilizing the cellular membrane.12a The emissive properties of [4]Cl opened a way to visualize this phenomenon. First, the cytotoxicity of [4]Cl was evaluated in vitro on non-small lung cancer cell-line (A549). Whereas – in accordance with the literature11 – [3]Cl showed no measurable toxicity even after incubation for 24 h (see ESI† part 4 for details), its amphiphilic analogue [4]Cl showed significant cytotoxicity after only one hour incubation, characterized by an effective concentration (EC50,1h) of 9.8 μM. This cytotoxic activity increased with incubation time, reaching an EC50,24h value of 2.2 μM after 24 h incubation, which was twice lower than that of cisplatin (EC50 = 4.3 μM). As [4]Cl is phosphorescent, the uptake could this time be followed by optical microscopy time-lapse imaging. The complex was taken up exceptionally quickly, i.e. after only a few minutes of incubation (Fig. 4). Time-lapse studies (Video S1, ESI†) showed that the cell membrane was initially stained, as expected due to the amphiphilic character of [4]Cl (Fig. 4). However, it also later internalized: after 9 h (Fig. S19, ESI†) bright emission could be detected in the perinuclear region, indicating that the molecule was enriched probably in the endoplasmic reticulum. Interestingly, the nucleus clearly showed no red emission, as observed by z-stacking (Video S2, ESI†), which indicates either that [4]Cl did not enter the nucleus, or that its emission there was quenched. As instead of stained nuclei spherical red organelles were observed after 9 h incubation, we hypothesize that interaction with nuclear DNA is not likely to explain the toxicity of [4]Cl, but that exosomes, lysosomes, or even mitochondrial membranes, incorporate the amphiphilic complex [4]Cl, and that the cytotoxicity is based on membrane modulation.
In summary, we report here on compound [4]Cl as a new type of amphiphilic phosphorescent [Ru(tpy)(bpy)(Cl)]+ analogue. By attaching an amide group R on the 4′-position of the terpyridine ligand, the 3MLCT level was stabilized just enough to switch on red emission without compromising photosubstitution of the monodentate ligand. By attaching a long alkyl tail to the positively charged ruthenium complex, compound [4]Cl was made amphiphilic and self-assembled in aqueous solutions. Because of the thermal hydrolysis of the Ru–Cl bond, self-assembly was found to depend on chloride concentration: nanometer-scale micelles of the aqua complex [7]2+ were found in pure water, while micrometer-scale aggregates of [4]+ were characterized in PBS. Whereas the reference compound [3]Cl is essentially not cytotoxic, the amphiphilicity of [4]Cl made it more toxic than cisplatin in A549 cancer cells. Most importantly, red emission now allows for probing cell uptake and localization of the complex by optical microscopy, which was impossible with all previously known ruthenium compounds combining a terpyridine and a bipyridine ligand. Although [4]Cl seemed at short incubation times to behave like a simple soap and to attack the cell membrane, longer incubation times revealed a deeper cellular uptake, no red emission in the nucleus, and accumulation of the compound in the membrane-rich, peri-nuclear region (probably the endoplasmic reticulum). Overall, amide functionalization in 4′ position on the terpyridine appears as a very efficient manner to turn on the emission of analogues of [3]Cl without switching off photosubstitution properties. By tuning the functional group attached to the amide, it will be possible to modulate not only the lipophilicity of the complex, as achieved here, but also to introduce cancer-targeting groups for example, which will be critical for the development of PACT complexes based on this type of complexes.
The European Research Council is acknowledged for a Starting Grant to S. B. NWO is acknowledged for a VIDI grant to S. B., a VICI grant and a Middelgroot investment grant to J. T. M. K. The COST action CM1105 is acknowledged for stimulating scientific discussions. Prof. E. Bouwman and Dr S. L. Hopkins are kindly acknowledged for support and scientific discussions, Dr A. Boyle for helping with TEM imaging, and D. Saarloos for synthetical work. Prof. B. Koster, Dr R. I. Koning, and Dr T. Sharp are acknowledged for support and access to the cryoTEM facility at the LUMC.
Notes and references
- F. H. Burstall, J. Chem. Soc., 1936, 173–175 RSC.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS; (b) W. W. Brandt, F. P. Dwyer and E. C. Gyarfas, Chem. Rev., 1954, 54, 959–1017 CrossRef CAS; (c) J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. Decola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS; (d) S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, 2007, vol. 280, pp. 117–214 Search PubMed.
- (a) M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC; (b) H.-W. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763–11773 CrossRef CAS PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed.
- (a) W. D. Cao, J. P. Ferrance, J. Demas and J. P. Landers, J. Am. Chem. Soc., 2006, 128, 7572–7578 CrossRef CAS PubMed; (b) M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC; (c) T. Joshi, V. Pierroz, S. Ferrari and G. Gasser, ChemMedChem, 2014, 9, 1419–1427 CrossRef CAS PubMed; (d) L. Xu, N.-J. Zhong, H.-L. Huang, Z.-H. Liang, Z.-Z. Li and Y.-J. Liu, Nucleosides, Nucleotides Nucleic Acids, 2012, 31, 575–591 CrossRef CAS PubMed; (e) L. M. Loftus, J. K. White, B. A. Albani, L. Kohler, J. J. Kodanko, R. P. Thummel, K. R. Dunbar and C. Turro, Chem. – Eur. J., 2016, 22, 3704–3708 CrossRef CAS PubMed; (f) G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2015, 282–283, 127–138 CrossRef CAS; (g) A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551–6562 RSC; (h) A. Martin, A. Byrne, C. Dolan, R. J. Forster and T. E. Keyes, Chem. Commun., 2015, 51, 15839–15841 RSC.
- A. J. Göttle, F. Alary, M. Boggio-Pasqua, I. M. Dixon, J.-L. Heully, A. Bahreman, S. H. C. Askes and S. Bonnet, Inorg. Chem., 2016, 55, 4448–4456 CrossRef PubMed.
- (a) J. Fan, S. Tysoe, T. C. Strekas, H. D. Gafney, N. Serpone and D. Lawless, J. Am. Chem. Soc., 1994, 116, 5343–5351 CrossRef CAS; (b) J. Van Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853–4858 CrossRef CAS.
- O. A. Borg, S. S. M. C. Godinho, M. J. Lundqvist, S. Lunell and P. Persson, J. Phys. Chem. A, 2008, 112, 4470–4476 CrossRef CAS PubMed.
- E. Jakubikova, W. Chen, D. M. Dattelbaum, F. N. Rein, R. C. Rocha, R. L. Martin and E. R. Batista, Inorg. Chem., 2009, 48, 10720–10725 CrossRef CAS PubMed.
- (a) J. D. Knoll, B. A. Albani and C. Turro, Chem. Commun., 2015, 51, 8777–8780 RSC; (b) R. E. Goldbach, I. Rodriguez-Garcia, J. H. van Lenthe, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2011, 17, 9924–9929 CrossRef CAS PubMed; (c) J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed.
- O. Novakova, J. Kasparkova, O. Vrana, P. M. van Vliet, J. Reedijk and V. Brabec, Biochemistry, 1995, 34, 12369–12378 CrossRef CAS PubMed.
- (a) B. Siewert, V. H. S. van Rixel, E. J. van Rooden, S. L. Hopkins, M. J. B. Moester, F. Ariese, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2016, 22, 10960–10968 CrossRef CAS PubMed; (b) L. N. Lameijer, S. L. Hopkins, T. G. Brevé, S. H. C. Askes and S. Bonnet, Chem. – Eur. J., 2016, 22, 18484–18491 CrossRef CAS PubMed.
- (a) C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC; (b) S. Bonnet, Comments Inorg. Chem., 2015, 35, 179–213 CrossRef CAS.
- E. Alessio, Eur. J. Inorg. Chem., 2016, 1549–1560 Search PubMed.
- (a) K. Heinze, K. Hempel and M. Beckmann, Eur. J. Inorg. Chem., 2006, 2040–2050 CrossRef CAS; (b) J. Dietrich, U. Thorenz, C. Förster and K. Heinze, Inorg. Chem., 2013, 52, 1248–1264 CrossRef CAS PubMed; (c) A. Breivogel, M. Meister, C. Förster, F. Laquai and K. Heinze, Chem. – Eur. J., 2013, 19, 13745–13760 CrossRef CAS PubMed; (d) A. Breivogel, C. Kreitner and K. Heinze, Eur. J. Inorg. Chem., 2014, 5468–5490 CrossRef CAS.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- (a) A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed; (b) M. Mauro, G. De Paoli, M. Otter, D. Donghi, G. D'Alfonso and L. De Cola, Dalton Trans., 2011, 40, 12106–12116 RSC.
- S. Yoon, P. Kukura, C. M. Stuart and R. A. Mathies, Mol. Phys., 2006, 104, 1275–1282 CrossRef CAS.
- F. Alary, J. L. Heully, L. Bijeire and P. Vicendo, Inorg. Chem., 2007, 46, 3154–3165 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans., 1976, 72, 1525–1568 RSC.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- (a) T. A. Wagay, J. Dey, S. Kumar, V. K. Aswal and K. Ismail, RSC Adv., 2016, 6, 66900–66910 RSC; (b) T. A. Wagay, J. Dey, S. Kumar, V. K. Aswal and K. Ismail, Colloids Surf., A, 2016, 503, 61–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Graphical results, and videos. CCDC 1534260. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc02989f |
| This journal is © The Royal Society of Chemistry 2017 |