
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intracellular delivery of a catalytic organometallic complex†
Eugenio
Indrigo‡
,
Jessica
Clavadetscher‡
,
Sunay V.
Chankeshwara
 ,
Alicia
Megia-Fernandez
,
Annamaria
Lilienkampf
and
Mark
Bradley
*
,
Alicia
Megia-Fernandez
,
Annamaria
Lilienkampf
and
Mark
Bradley
*
EaStCHEM, School of Chemistry, University of Edinburgh, David Brewster Road, EH9 3FJ Edinburgh, UK. E-mail: Mark.Bradley@ed.ac.uk
First published on 6th June 2017
Abstract
A homogeneous carbene-based palladium catalyst was conjugated to a cell-penetrating peptide, allowing intracellular delivery of catalytically active Pd complexes that demonstrated bioorthogonal activation of a profluorophore within prostate cancer cells.
In recent years, bioorthogonal organometallic reactions have been successfully applied in chemical biology.1–8 In particular, Pd-mediated chemical transformations have been used to modify or synthesise molecules through cleavage of protecting groups or cross-coupling reactions.5,9 The first use of Pd chemistry in mammalian cells was reported by Bradley,10 who trapped Pd nanoparticles within an inert polymer microsphere for intracellular delivery. This “active” transition metal catalyst was used to mediate C–C bond formation via Suzuki–Miyaura cross-coupling and allyl carbamate cleavage reactions inside living cells, including the intracellular activation of a prodrug of the antineoplastic agent amsacrine. This approach was also used to convert propargyl derivatives of 5-fluorouracil and gemcitabine to their active forms, as well as catalyse Suzuki–Miyaura cross-couplings of drugs in the presence of an extracellular heterogeneous Pd catalyst.11–13 Pd-mediated transformations have since been used to selectively activate biomolecules in cells. Chen developed the chemical activation of proteins based on the decaging of propargylcarbamate caged lysine with simple Pd salts, while Pd(NO3)2, Pd(dba)2, and allyl2PdCl2 have also been used to mediate Sonogashira cross-coupling reactions inside bacteria,14 or to activate a protein of interest inside cells.14–17 However, under optimised conditions, 50 equivalents of these Pd complexes were required for the completion of the cross-coupling reactions.16 The major drawbacks of all the above methods are the lack of cell selectivity of the catalyst and the use of stoichiometric, or even excess (up to 500 eq.), Pd complex and the cellular toxicity when used in non-complexed/encapsulated form.5,18 Targeted localisation of a Pd catalyst, by means of cell selective delivery of a homogeneous catalyst, would represent a major advance in the field.
Herein, we have extended the scope of Pd catalysed chemistry in living systems by developing a method to deliver and track an active organometallic complex inside mammalian cells. These water-soluble and traceable catalysts were based on a Pd(II)–carbene complex coupled to a fluorescently labelled homing peptide for targeted cell delivery. The biocompatible homogeneous Pd catalyst was based on a previously reported carbene Pd ligand coupled to a peptide.19,20 N-Heterocyclic carbenes (NHC) are strong σ-electron donating molecules (enhancing the rate of oxidative addition), coordinate tightly to Pd centres (disfavouring aggregation), and can have bulky substituents (increasing the rate of reductive elimination).21,22 Peptides were chosen to deliver the Pd complex inside cells as they are biocompatible, easy to prepare by solid-phase peptide synthesis (SPPS), versatile, readily labelled and functionalised, with known cell delivery abilities.23
The polycationic (tri-lysine) catalyst 1 represented a simple cell penetrating peptide (CPP) which was able to cross the cell membrane,23 and could be readily labelled to allow tracking of the catalyst inside cells.
Here, either 5(6)-carboxyfluorescein (FAM, λex/em = 492/517 nm) or sulfonated Cy5 (λex/em = 630/654 nm) were attached to the peptides to give the fluorescent Pd-catalysts 2 and 3, respectively (Scheme 1). Peptides 1, 2 and 3 were synthesised on a Rink amide linker functionalised aminomethyl polystyrene resin using standard Fmoc/tBu SPPS with Oxyma/DIC as the coupling combination (Scheme 1 and ESI†). Coupling of the fluorophores in 2 and 3 was achieved by introducing a bis-N-[1-(4,4-dimethyl-2,6-dioxocyclo hexylidene) ethyl] (Dde) protected Lys as the first amino acid (Fmoc-Lys(Dde)-OH). Prior to Fmoc removal of the terminal Lys residue, the Dde group was orthogonally removed with hydroxylamine24 and the liberated amine coupled to the activated dye. The imidazole ligand 4 was coupled to the amino terminus of the peptides using TBTU as an activating agent. The imidazole salt was treated with the base BEMP to form the carbene, which was subsequently trapped by the addition of dichloro(1,5-cyclooctadiene) palladium(II) (PdCl2COD) as the source of Pd.19 The catalysts were cleaved from the resin by treatment with TFA (5% water, addition of conventional scavenging agents destroyed the Pd complex) and purified by preparative HPLC. To evaluate the catalytic activity in a biological setting, non-fluorescent bis-propargyloxycarbonyl (Proc) rhodamine 110 (5) was treated with Pd catalyst 1 in phosphate buffered saline (PBS) and PC-3 (prostate adenocarcinoma) cell lysate (see ESI†). Upon Pd catalysed cleavage of the protecting groups with 0.1 eq. of 1 for 24 h, fluorescent 6 was generated giving a 200-fold increase in fluorescence. The reaction in cell lysate resulted in a more modest 9-fold relative increase in fluorescence with 0.1 eq. of the catalyst, but demonstrated that the Pd–peptide hybrid was catalytically competent even in the presence of a complex mixture of biomolecules.
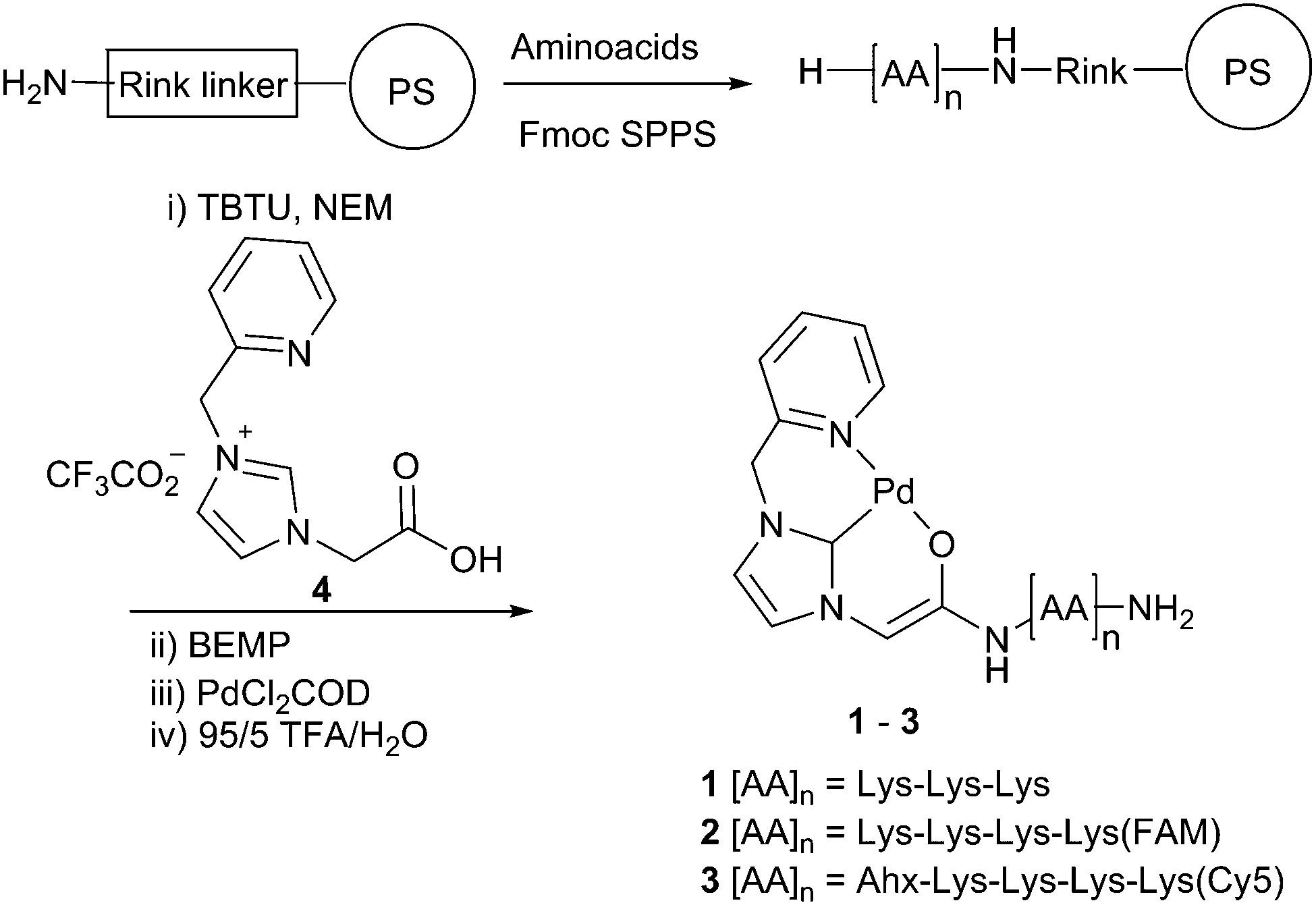 | ||
| Scheme 1 Synthetic route to the Pd–peptide catalysts on a Rink functionalised polystyrene resin. | ||
The cellular uptake of the carboxyfluorescein labelled Pd–peptide 2 was investigated with the total quantity of the internalised Pd determined by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis of PC-3 cells (Fig. 1B), giving a total Pd content of 20 ng per million cells (when incubated with 30 μM of 2 for 2 h), showing a higher metal uptake compared to cells treated with 10 μM of Pd(OAc)2 (higher concentration of Pd(OAc)2 showed cytotoxicity). Catalyst 2 showed no cytotoxicity even at 200 μM (see ESI†). The cellular uptake and intracellular location of the Pd catalyst 2 was investigated by flow cytometry and confocal microscopy (Fig. 1C and D). PC-3 cells showed a shift of the whole cell population towards higher fluorescence intensity after 2 h incubation with catalyst 2 (30 μM), with the fluorescence localised within the cytoplasm and, unexpectedly, in the nucleus (cationic peptides generally localise in the cytoplasm and in vesicles25,26).
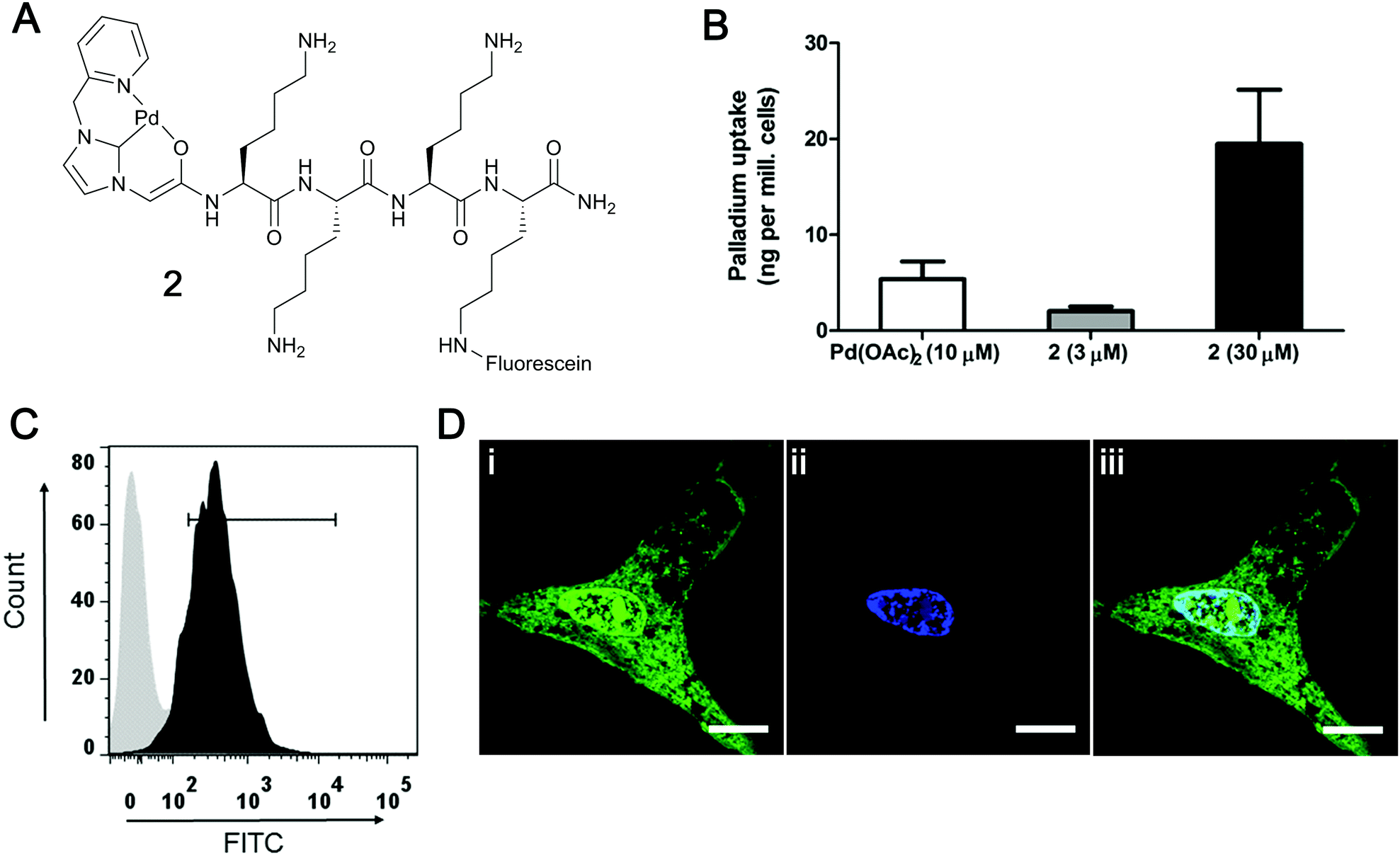 | ||
| Fig. 1 Cellular uptake of Pd–peptide 2. (A) Structure of the carboxyfluorescein labelled Pd–peptide 2. (B) Intracellular Pd content (ng Pd per million cells) of PC-3 cells incubated with 10 μM Pd(OAc)2 or 2 (3 μM and 30 μM) for 2 h (higher concentrations of Pd(OAc)2 were toxic). Cells were harvested, lysed and the cell content was subjected to ICP-OES measurements. (C) Flow cytometry histograms showing the uptake of 2 (30 μM) in PC-3 cells after 2 h (in black) and untreated control cells (in grey). (D) PC-3 cells were incubated with 2 (30 μM) for 2 h, fixed with 4% paraformaldehyde, stained with DAPI (nuclei stain) and imaged by confocal microscopy. Panels show (i) Pd catalyst 2 (green, λex = 488 nm) located in the cytoplasm and nucleus, (ii) cell nucleus (blue, λex = 405 nm), and (iii) merged image (light blue indicates co-localisation). Scale bar 20 μm. | ||
The catalytic activity of the Pd–peptide catalysts in cell-based assays via Proc-rhodamine 5 activation was investigated with the Cy5 labelled Pd–peptide 3. PC-3 cells were incubated with 3 (30 μM) for 2 h, washed to remove any extracellular catalyst, and subsequently incubated with 5 (50 μM) for 18 h (Fig. 2A). Fluorescence microscopy verified the presence of the Pd-catalyst 3 as well as synthesised 6 within the cytoplasm of PC-3 cells (Fig. 2B). Analysis by flow cytometry showed a shift of the whole cell population towards higher fluorescence intensity in the Cy5 channel indicating the uptake of the catalyst, while a shift to higher FITC intensity indicated the formation of 6 (Fig. 2C).
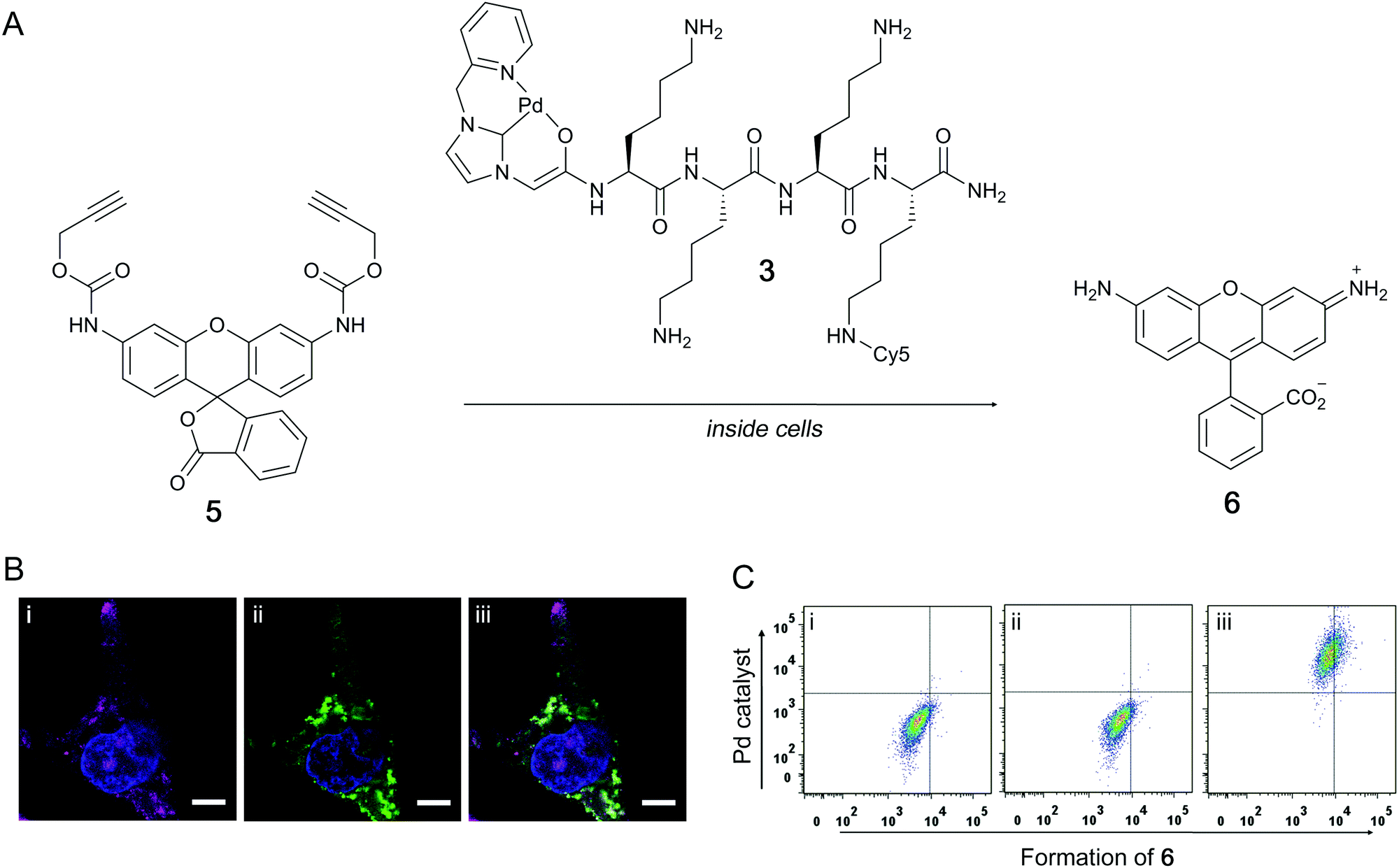 | ||
| Fig. 2 Pd catalysed decaging of Proc-rhodamine 5 in PC-3 cells. (A) Catalysed fluorescence “switch-on” of the caged fluorophore 5 with the Pd–peptide complex 3. Cells were incubated with Pd–peptide 3 (30 μM) for 2 h, followed by incubation with 5 (50 μM) for 18 h. (B) Confocal microscopy images of PC-3 cells fixed with 4% paraformaldehyde and stained with DAPI (nuclei stain). Panels show: (i) cell nucleus (blue, λex = 405 nm) and Pd–peptide 3 (magenta, λex = 633 nm), (ii) cell nucleus (blue) and in situ synthesised 6 (green, λex = 488 nm), and (iii) merged image. Scale bar 20 μm. (C) Flow cytometry cytograms showing: (i) untreated control cells, (ii) cells treated with Proc-rhodamine 5, and (iii) cells treated with 5 and 3. | ||
In conclusion, we have demonstrated the ability to combine a catalytically active Pd complex with a cell-delivery peptide scaffold, which was able to catalyse a depropargylation reaction in a biologically relevant setting. The non-cytotoxic Pd catalyst was successfully used in the fluorescent labelling of mammalian cells, showing significant catalytic activity in an intracellular environment. Future applications will thus include the activation of prodrugs via these Pd catalysts. This new type of catalytic system gives the basis for a wide range of biocompatible catalysts, where their facile modification allows the incorporation of a variety of different targeting peptides that could selectively enter different cells or specific tissues.
Notes and references
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- J. Li and P. R. Chen, ChemBioChem, 2012, 13, 1728–1731 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- S. V. Chankeshwara, E. Indrigo and M. Bradley, Curr. Opin. Chem. Biol., 2014, 21C, 128–135 CrossRef PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 Search PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212–14214 RSC.
- J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330–7338 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- N. Li, R. K. V. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Chem. Commun., 2013, 49, 2747–2749 RSC.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- K. Worm-Leonhard and M. Meldal, Eur. J. Org. Chem., 2008, 2008, 5244–5253 CrossRef.
- J. F. Jensen, K. Worm-Leonhard and M. Meldal, Eur. J. Org. Chem., 2008, 2008, 3785–3797 Search PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 CrossRef CAS PubMed.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, Cell. Mol. Life Sci., 2005, 62, 1839–1849 CrossRef CAS PubMed.
- J. J. Díaz-Mochón, L. Bialy and M. Bradley, Org. Lett., 2004, 6, 1127–1129 CrossRef PubMed.
- R. Fischer, K. Köhler, M. Fotin-Mleczek and R. Brock, J. Biol. Chem., 2004, 279, 12625–12635 CrossRef CAS PubMed.
- G. Drin, S. Cottin, E. Blanc, A. R. Rees and J. Temsamani, J. Biol. Chem., 2003, 278, 31192–31201 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Proc-rhodamine decaging and biological studies. See DOI: 10.1039/c7cc02988h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |