
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the chemical transformations of functionalized alkylidenecyclopropanes (FACPs)
Liu-Zhu
Yu
a,
Kai
Chen
a,
Zi-Zhong
Zhu
a and
Min
Shi
 *ab
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, P. R. China. E-mail: mshi@mail.sioc.ac.cn
bKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 MeiLong Road, Shanghai 200237, P. R. China
First published on 8th May 2017
Abstract
During the past several years, functionalized alkylidenecyclopropanes (FACPs) have attracted intensive attention in synthetic chemistry. Many interesting transformations of FACPs have been developed to synthesize a lot of structurally diverse and valuable polycyclic and heterocyclic compounds. This review will classify FACPs into aryl-FACPs, alkyl-FACPs and ring-FACPs for the first time, and recent interesting chemical transformations in these research fields will be included, respectively, from 2011. Moreover, we will pay more attention to the clarification of the reaction mechanism, in which the C–C bond cleavage of alkylidenecyclopropanes (ACPs) will be emphasized.
 Liu-Zhu Yu | Liu-Zhu Yu was born in 1989 in Anhui (P. R. China). He received his BS degree in 2013 from the East China University of Science & Technology, before joining a PhD program in the same university. He is currently a PhD candidate under the guidance of Professor Min Shi. |
 Kai Chen | Kai Chen was born in 1989 in Jiangsu (P. R. China). He received his BS degree in 2011 from East China University of Science & Technology, before joining a PhD program in the same university. He is currently a PhD candidate under the guidance of Professor Min Shi. |
 Zi-Zhong Zhu | Zi-Zhong Zhu was born in 1989 in Shandong (P. R. China). He received his BS degree in 2013 from the Qingdao University of Science & Technology, before joining a MS program in the East China University of Science & Technology. He is currently a Master's degree candidate under the guidance of Professor Min Shi. |
 Min Shi | Dr Min Shi was born in 1963 in Shanghai, China. He received his BS in 1984 (Institute of Chemical Engineering of East China) and PhD in 1991 (Osaka University, Japan). He had his postdoctoral research experience with Prof. Kenneth M. Nicholas at the University of Oklahoma (1995–1996) and worked as an ERATO Researcher in Japan Science and Technology Corporation (JST) (1996–1998). He is currently a group leader of the State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC). |
1. Introduction
Strained small carbocycles have long been the concern of organic chemists. In particular, among them, the cyclopropane subunit plays a prominent role in organic chemistry.1 In the class of cyclopropanes, methylene- or alkylidenecyclopropanes (MCPs or ACPs) as highly strained but readily accessible molecules can undergo a variety of ring-opening reactions because the relief of ring strain can provide a potent thermodynamic driving force.2 These ring-opening processes can trigger various reactions of MCPs or ACPs with other substrates, giving efficient access to enhanced molecular complexity in organic syntheses. Generally, the reactivity of MCPs or ACPs has been fully excavated and it includes transition metal-catalyzed reactions,3 Lewis acid-catalyzed reactions,4 free radical addition induced rearrangements,5 thermal induced cyclizations6 and other types.7In recent years, the development of MCPs or ACPs has emerged as a new direction using novel functionalized alkylidenecyclopropanes (FACPs) as substrates, and many interesting reactions have been explored with these novel FACPs by several groups. These novel FACPs include three types, namely, aryl-functionalized alkylidenecyclopropanes (aryl-FACPs), alkyl-functionalized alkylidenecyclopropanes (alkyl-FACPs) and ring-functionalized alkylidenecyclopropanes (ring-FACPs). Transformations of them to interesting and useful polycyclic compounds effectively expand this field of strained small carbocycles and it is necessary for further exploration to use these transformations to realize the total synthesis of natural products (Scheme 1).
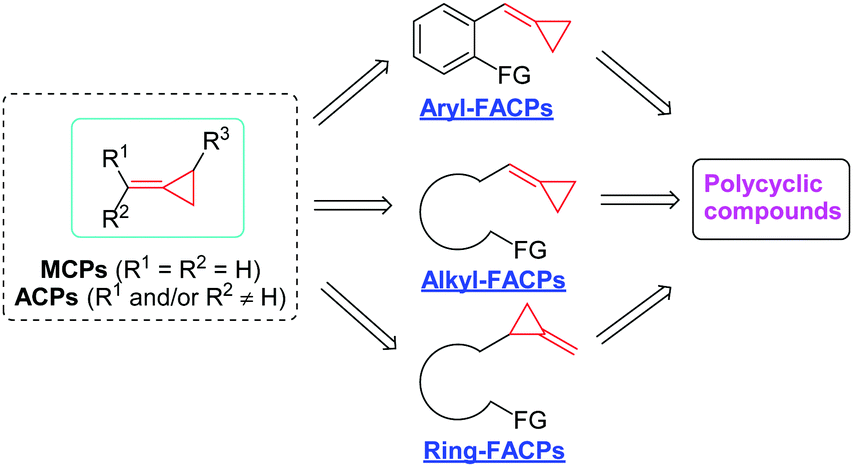 | ||
| Scheme 1 Classification of functionalized alkylidenecyclopropanes (FACPs). | ||
Recently, several excellent reviews on this fascinating area with regard to the use of MCPs or ACPs in organic synthesis have been published.2 For example, in 2014, Brandi, Pellissier and Yu summarized the recent progress in this field as a review article, respectively.2k,l,n However, a focused review of the chemical transformation of FACPs has never been reported before. This minireview will put the concept of FACPs forward for the first time and mainly focuses on the recent advances in the chemical transformations of FACPs by our group (Scheme 2), along with other groups' findings from 2011. We hope this minireview will satisfy the expectations of the readers who are interested in the field of novel chemical transformation of FACPs.
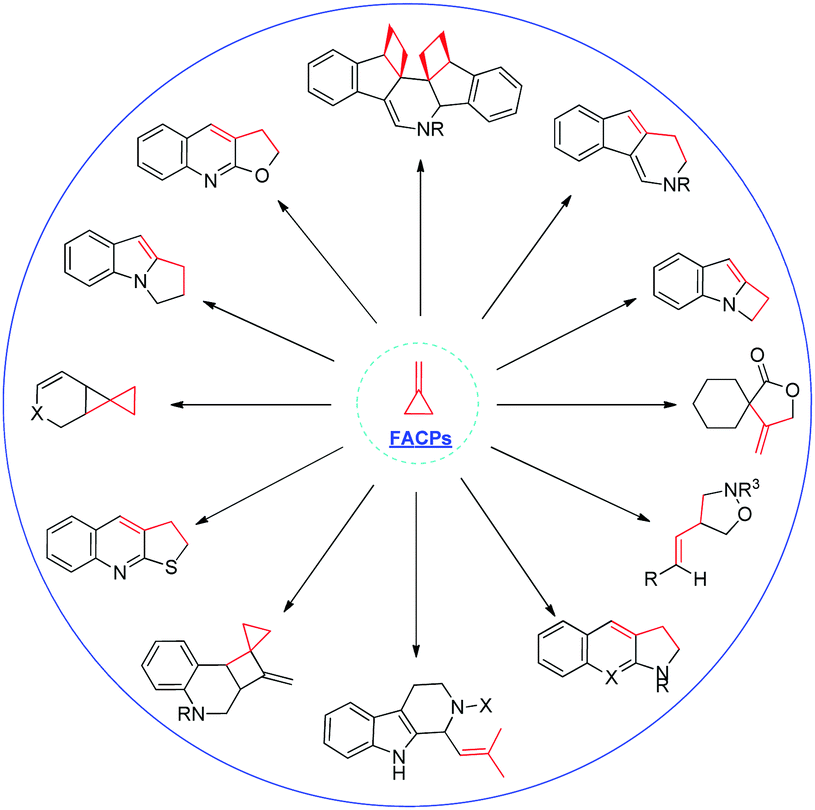 | ||
| Scheme 2 Represented examples reported by our group. | ||
2. Reactivity of FACPs
2.1 Transformations of aryl-FACPs
In early 2005, Yamamoto's group reported the first Pd0-catalyzed reaction that transforms aniline-tethered alkylidenecyclopropanes 1 to six-membered exomethylene nitrogen heterocycles 2 in good yields (Scheme 3).8 This reaction starts with the intramolecular oxidative addition of a nitrogen–hydrogen bond of amine onto a zero valent palladium species to produce an amino-palladium species 3, followed by hydropalladation of alkylidenecyclopropane to give 4. The cyclopropylpalladium species 4 most probably undergoes β-carbon elimination, leading to an allylpalladium intermediate 5. Subsequent reductive elimination of Pd(II) gives product 2.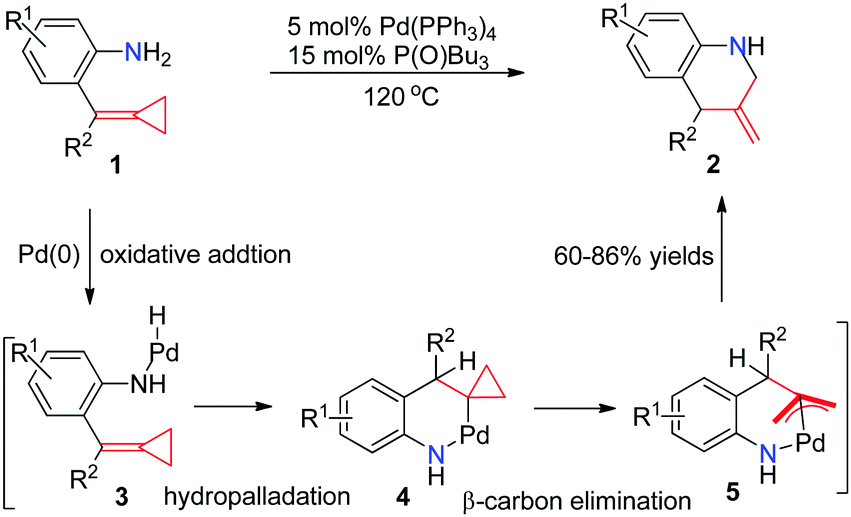 | ||
| Scheme 3 Palladium-catalyzed reaction of FACPs 1. | ||
Yamamoto's work greatly inspired us to do further research on these kinds of FACPs. In 2012, we reported a convenient synthetic method to obtain functionalized pyrrolo[1,2-a]indoles 6 in good yields, which are common core structures found in a number of naturally occurring compounds via a ring-opening reaction and thermal-induced cyclization from aldehydes with aniline-tethered ACPs 1 (Scheme 4).9 The reaction of 1 with benzaldehyde in the presence of anhydrous magnesium sulfate afforded imine 7. When it was heated in toluene at 110 °C, thermal induced [3+2] cyclization took place, leading to the formation of 6 through a three-membered ring-opening pathway.
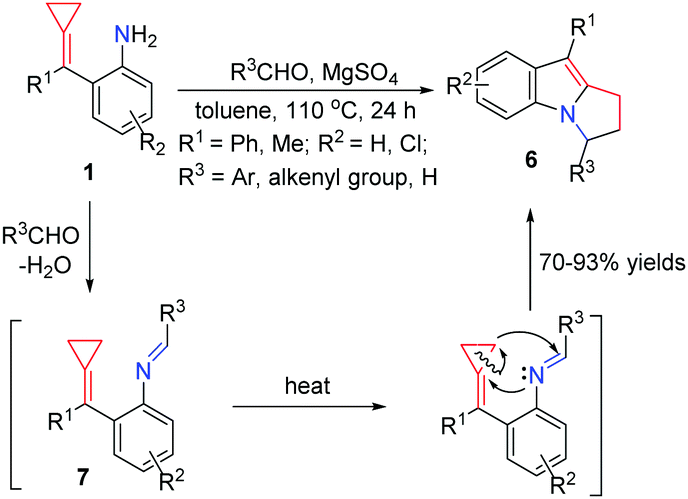 | ||
| Scheme 4 Thermal-induced cyclization from aldehydes with FACPs 1. | ||
More recently, for more reaction partners to realize thermal induced [3+2] cyclizations of aniline-tethered ACPs 1, our group developed a facile method to access a range of furoquinoline and thienoquinoline derivatives 8 and 11via in situ generated isocyanate and isothiocyanate intermediates (Schemes 5 and 6).10 These reactions produced the desired products in excellent isolated yields and tolerated a broad substrate scope, thus providing a potential application to synthesize some related natural products and medical candidates. What is more, the production of both 8 and 11 is easy to scale-up to gram scale without loss of reaction efficiency. We proposed a reasonable mechanism, as outlined in Schemes 5 and 6, respectively. It is a common step that isocyanate-tethered ACPs 9 or isothiocyanate-tethered ACPs 12 were first formed based on the general methods. Subsequently, a consecutive 6π-electrocyclization and rearrangement occurred to produce the corresponding furoquinoline and thienoquinoline derivatives 8 and 11.
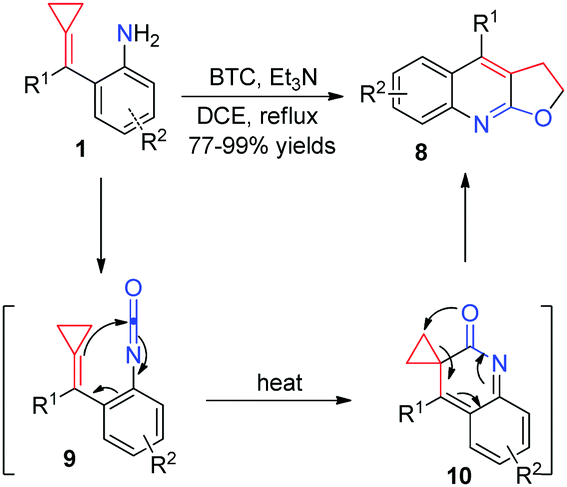 | ||
| Scheme 5 Formation of furoquinoline derivatives from FACPs 1. | ||
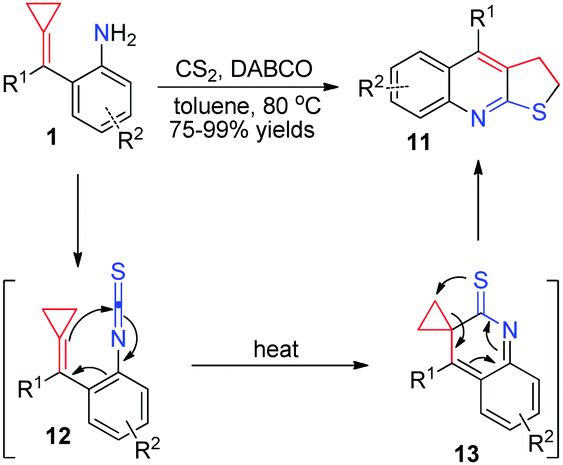 | ||
| Scheme 6 Formation of thienoquinoline derivatives from FACPs 1. | ||
For different types of thermal induced cyclization and cycloaddition of FACPs, in 2013, we reported a facile synthetic method for bicyclo[4.2.0] nitrogen heterocycles 15via a thermal induced intramolecular [2+2] cycloaddition reaction of allene-ACPs 14 (Scheme 7).11 The DFT calculations indicate that this intramolecular [2+2] cycloaddition proceeds via a concerted manner and the strained small ring is necessary.
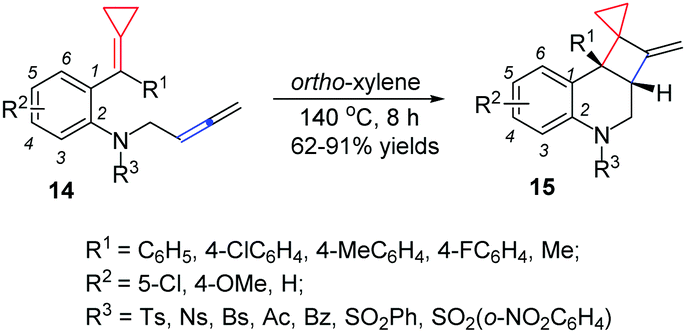 | ||
| Scheme 7 Thermal induced intramolecular [2+2] cycloaddition of 14. | ||
The cyclobutane group in the obtained bicyclo[4.2.0] nitrogen heterocycles 15 can be converted into a cyclobutene group along with the ring opening of cyclopropane. For example, upon treatment of BiCl3 in toluene at 80 °C for 10 h, product 15a could be easily transformed into product 16 in 78% yield, which is a novel type of 2a,3,4,8b-tetrahydrocyclobuta[c]quinoline derivative that has not been intensively studied so far (Scheme 8).
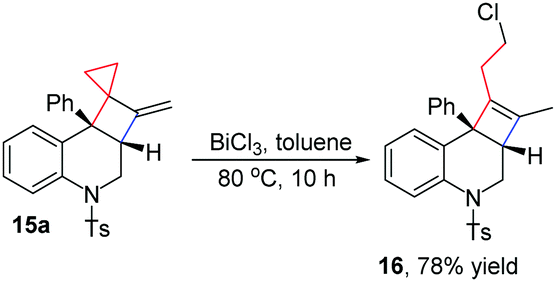 | ||
| Scheme 8 Transformation of compound 15a to 16. | ||
Recently, our group focused on acrylamide-tethered alkylidenecyclopropanes 17 and disclosed a novel iron or copper catalyzed trifluoromethylation of them with Togni reagent II 18 to construct two types of CF3-containing tetracyclic benzazepine derivatives 19 and 20 depending on whether or not R1 is a para-methoxy-substituent (Scheme 9).12 The reaction exhibited high chemoselectivity in which the CF3 radical preferentially attacked the acrylamide moiety followed by a ring-opening process of ACP. Most functional groups were tolerated, providing the corresponding benzazepine derivatives 19 and 20 in moderate to good yields. It is to be noted that, for the formation of 19, the electronic effect had a prominent impact on the reaction outcome. Thus, the reactions were carried out with Cu(MeCN)4PF6 as a catalyst and MeCN as the solvent to improve the reaction efficiency and deliver the desired products cleanly when the benzene ring was substituted by a strongly electron-donating methoxyl group. Products 20 were formed in moderate yields in the presence of a catalytic amount of FeCl2 in MeOH within 1 h.
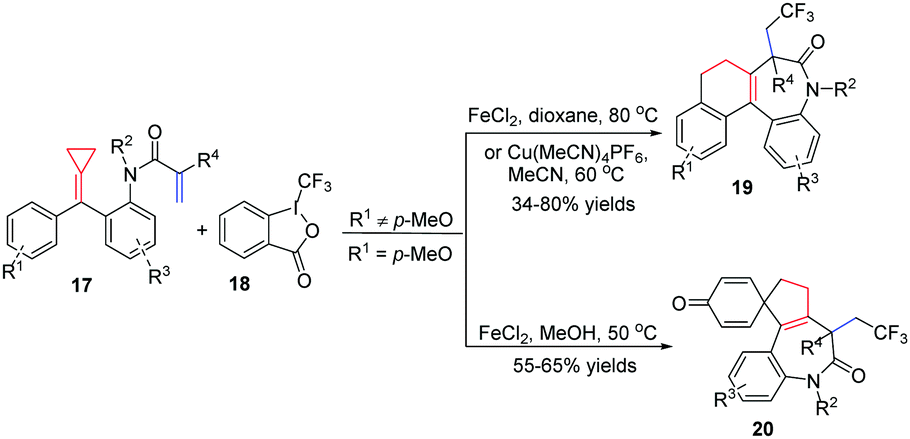 | ||
| Scheme 9 Iron or copper-catalyzed trifluoromethylation of FACPs 17. | ||
The reaction pathway is proposed in Scheme 10 based on radical trapping experiments and related precedents on trifluoromethylation of activated alkenes. The CF3 radical generated via a single-electron transfer (SET) process reacts chemoselectively with the acrylic moiety of FACPs 17 to give a radical intermediate 21. Subsequently, radical addition and ring-opening of alkylidenecyclopropane take place, producing homoallyl radical intermediate 23. Then, intramolecular cyclization of radical intermediate 23 and oxidation deliver product 19. However, if R1 is a para-methoxy-substituent, an ipso-cyclization occurs forming radical intermediate 25. Thus, the corresponding spirocyclic benzazepine 20 is obtained after oxidation.
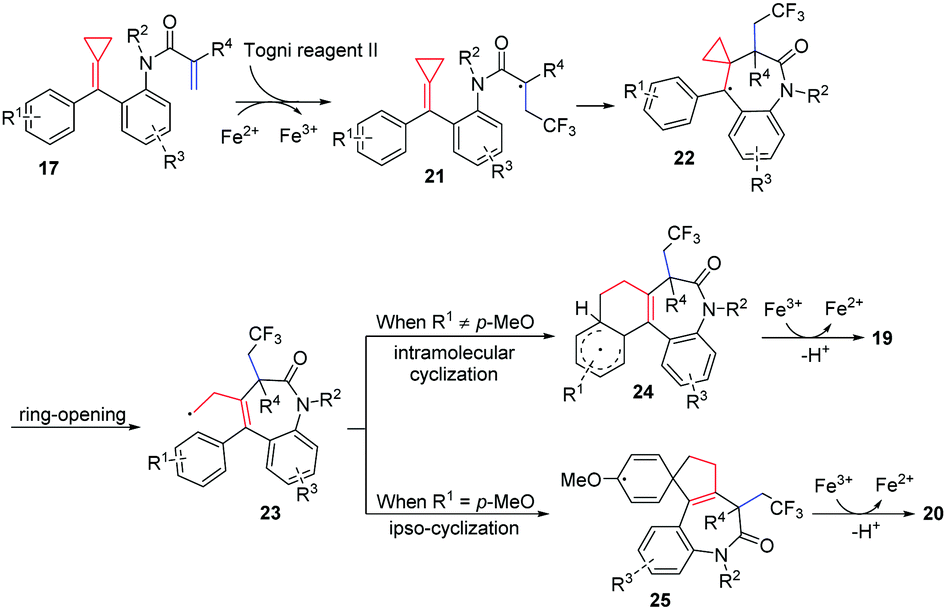 | ||
| Scheme 10 Proposed mechanism for the formation of 19 and 20. | ||
In 2011, Zhang and co-workers developed a new Ni0-catalyzed [3+2] cycloaddition of ACPs 26 with intramolecular aryl alkynes by the cleavage of a proximal C–C bond to obtain cyclopenta[a]indene derivatives 27 in good yields (Scheme 11).13 A plausible mechanism for the reaction is shown in Scheme 11. Oxidative addition of nickel(0) to the proximal C–C σ bond of cyclopropyl alkene leads to the nickelacyclobutane intermediate 29. The subsequent intramolecular addition of species 29 into the C–C triple bond generates the six-membered nickel cycle 30, which undergoes reductive elimination to give the corresponding product 27.
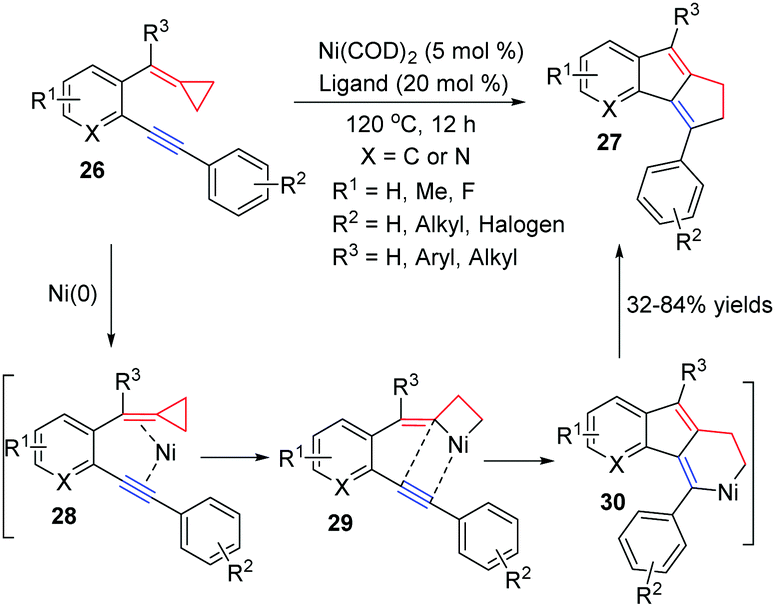 | ||
| Scheme 11 Nickel-catalyzed cycloaddition of FACPs 26. | ||
In 2011, Wu's group discovered a novel CuI-catalyzed cascade reaction of 2-ethynylaryl ACPs 31 with sulfonyl azides to generate fused indolines 32 in moderate to good yields (Scheme 12).14 They reasoned that 2-ethynylaryl ACPs 31 would react with sulfonyl azide catalyzed by CuI to afford the triazole intermediates 33. This intermediate could be transformed into the reactive ketenimines 34via a ring-opening rearrangement. A consecutive 6π-electrocyclization would take place to form intermediates 35, which subsequently underwent a rearrangement to give the desired products 32.
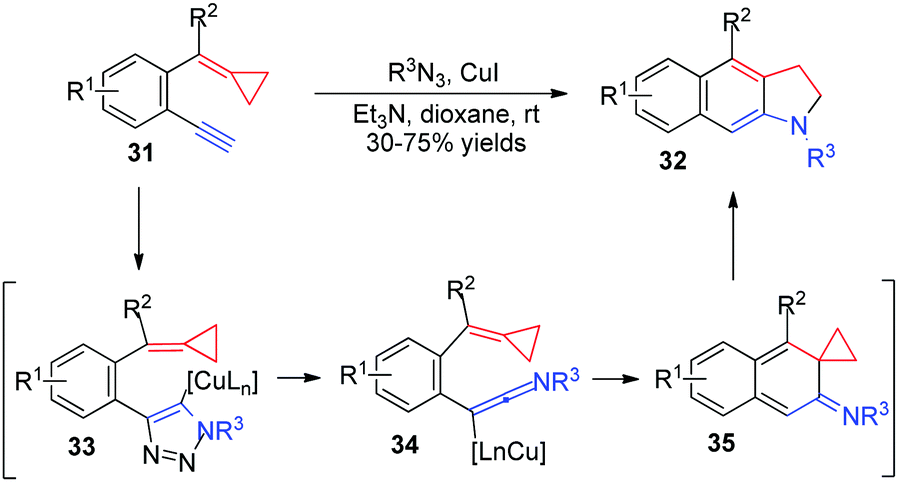 | ||
| Scheme 12 CuI-catalyzed cascade reaction of FACPs 31. | ||
In 2012, Wu's group reported another CuII-catalyzed tandem reaction of 1-bromoethynyl-2-(cyclopropylidenemethyl)arenes 36 with N-allylsulfonamide 37 to afford functionalized benzoindolines 38 in good yields (Scheme 13).15 The transformation may be a four-step cascade involving Ullmann coupling to get 39, aza-Claisen rearrangement to form 40, 6π-electrocyclization to obtain 41 and intramolecular rearrangement to produce benzoindolines 38.
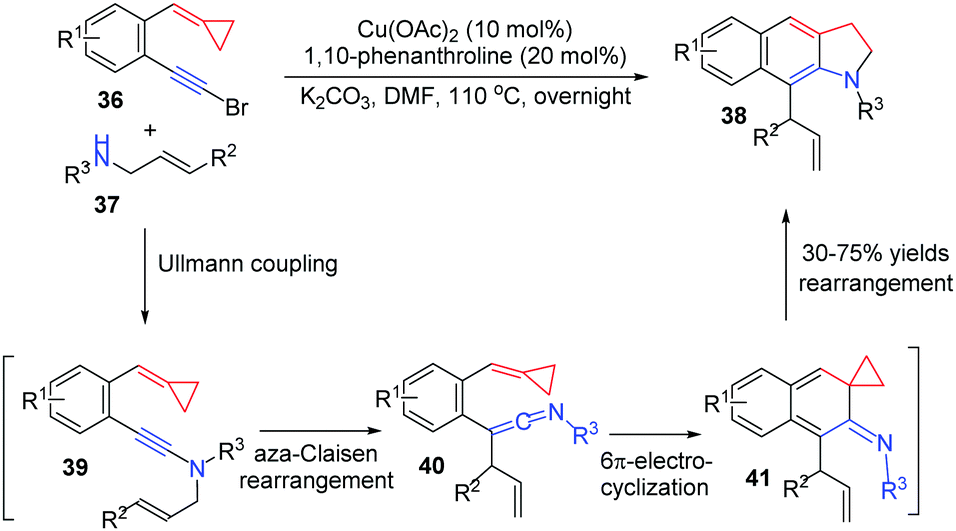 | ||
| Scheme 13 CuII-catalyzed cascade reaction of FACPs 36 with 37. | ||
Transition metal catalyzed intermolecular reactions of carbenes or nitrenes with MCPs have drawn great attention during the past decade. In 2014, we reported a RhII-catalyzed novel cascade cycloisomerization/aza-Diels–Alder reaction of 1-(cyclopropylidenemethyl)-2-(N-sulfonyltriazole)arenes 42, producing a series of highly functionalized polycyclic N-heterocycles 43 in moderate to good yields (Scheme 14).16
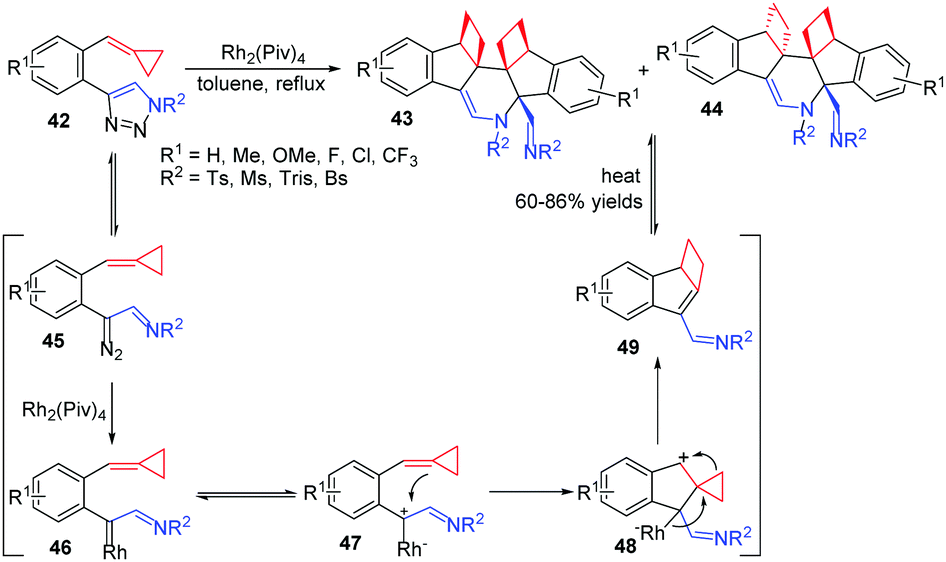 | ||
| Scheme 14 RhII-Catalyzed novel cascade reaction of FACPs 42. | ||
In this reaction, N-sulfonyltriazole 42 exists in equilibrium with its diazoimine tautomer 45. It can be efficiently intercepted by a RhII catalyst to give rise to highly reactive rhodium(II) azavinyl carbene 46. This intermediate exists in equilibrium with its diazoimine tautomer 47, followed by a nucleophilic attack from ACP furnishing intermediate 48. Subsequent ring expansion and elimination of the RhII catalyst generates the α,β-unsaturated imine intermediate 49, which is very reactive to induce an intermolecular aza-Diels–Alder [4+2] reaction to give products 43 and 44. When the product mixture is heated at 110 °C, the thermodynamically more stable product 43 can be isolated as the sole product because of the retro-Diels–Alder reaction process.
Next, we aimed to demonstrate the divergent reaction pathways of ACPs with azavinyl carbenes. Thiophene and cycloalkene tethered-triazole-ACPs 50 and 52 were synthesized and treated under standard conditions, only affording the corresponding fused indolines 51 and 53 in 73% and 55% yields, respectively (Scheme 15, eqn (1) and (2)). It is possible that thermal-induced rearrangements of special substrates 50 and 52 are faster than the generation of their rhodium(II) azavinyl carbene intermediates. As for substrate 54, which has a phenyl substituent on the ACP moiety, formal [3+3] cyclization took place to give product 55 in 65% yield (Scheme 15, eqn (3)).
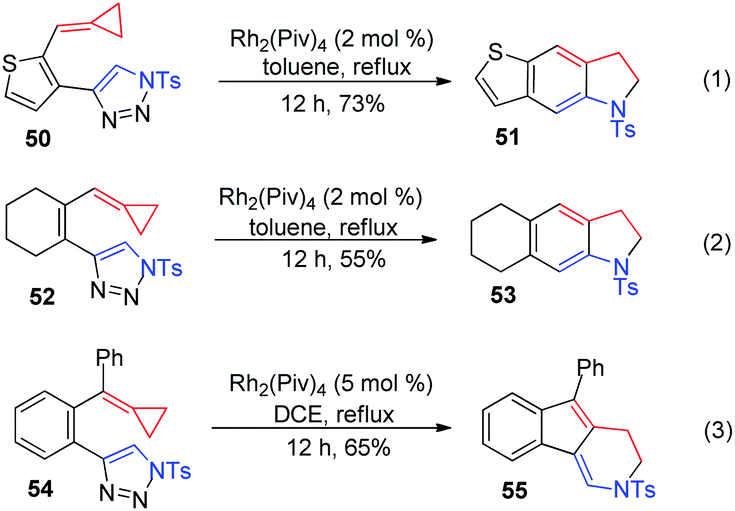 | ||
| Scheme 15 Further substrate scope study. | ||
The imino group of product 43a could be easily converted to amine 56 upon reduction with NaBH4 in methanol. To our delight, upon treatment of 56 with I2 and K2CO3, 3,8-diazabicyclo[3.2.1]octane derivative 57 could be obtained in 70% yield. This interesting reaction pathway may contain a tandem iodoamination, retro-Mannich reaction and hydrolysis (Scheme 16). Significantly, 3,8-diazabicyclo[3.2.1]octane derivatives usually have valuable biological properties. For example, azaprocin, which has the same core structure as that of 57, is an opioid analgesic with approximately ten times (10×) the potency of morphine, and a fast onset and short duration of action.
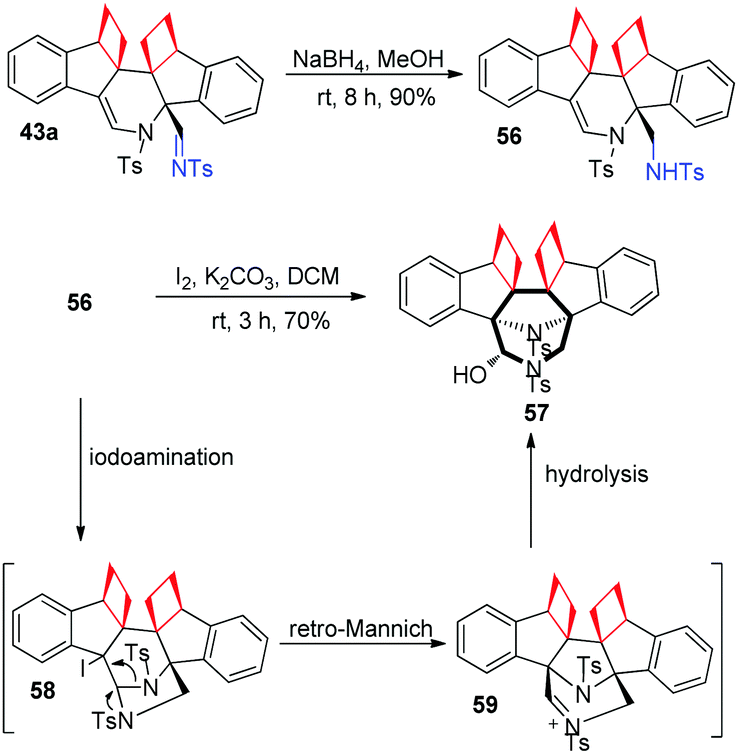 | ||
| Scheme 16 Transformations of compound 43a to 56 and 57. | ||
The reaction of nitrenes with MCPs should be very attractive because the incorporation of a nitrogen atom can potentially give rise to nitrogen containing heterocycles. However, according to the previously reported results, the intermolecular and intramolecular reactions of carbenes or nitrenes with MCPs all lead to the ring expansion of cyclopropane to cyclobutane. Recently, we disclosed a RhII-catalyzed intramolecular rearrangement of azide-ACPs 60 to form a series of indole-fused azetidines 61 in good yields (Scheme 17).17
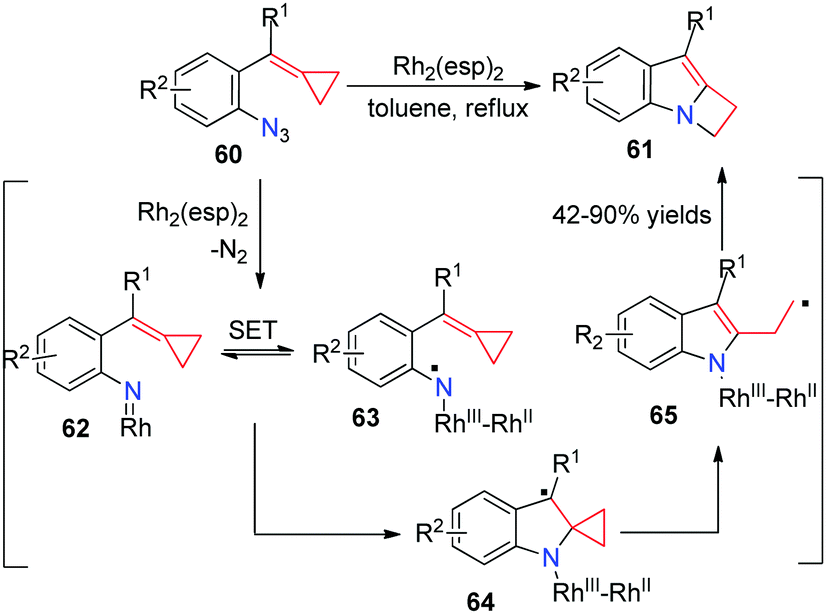 | ||
| Scheme 17 RhII-Catalyzed intramolecular rearrangement of azide-ACPs 60. | ||
A plausible mechanism is also outlined in Scheme 17. Coordination of the azide to Rh2(esp)2 and extrusion of N2 give the Rh-nitrene intermediate 62. Next, intramolecular single electron transfer (SET) takes place to form the N-centered radical species 63. The following radical addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the MCP moiety forms intermediate 64. It undergoes a ring-opening process to furnish homoallylic radical 65. Subsequent SET from the radical to RhIII and ring-closure gives rise to 61 together with the regeneration of the RhII catalyst. DFT calculations on the key step explain that cyclization and SET pathways are controlled by a radical clock in intermediate 64.
C bond in the MCP moiety forms intermediate 64. It undergoes a ring-opening process to furnish homoallylic radical 65. Subsequent SET from the radical to RhIII and ring-closure gives rise to 61 together with the regeneration of the RhII catalyst. DFT calculations on the key step explain that cyclization and SET pathways are controlled by a radical clock in intermediate 64.
The active radical intermediate 64 led us to question what would happen instead of ring-opening when there was no radical clock. Different ring sized azide-methylenecycloalkanes as well as dimethyl substituted alkenes were investigated. These reactions went on smoothly in chlorobenzene under reflux for 12 h to obtain a series of 3H-indoles in moderate to good yields. The reactions of methylenecyclobutanes 66 proceeded smoothly, affording hexahydrocyclopenta[b]indoles 67 in moderate yields after reduction by NaBH3CN (Scheme 18, eqn (1)). As for substrates 68, the corresponding tetrahydro-1H-carbazoles 69 were obtained in 62–80% yields (Scheme 18, eqn (2)). Specifically, in the case of methylenecyclohexanes 70, the reactions proceeded smoothly and the desired products were easily oxidized by air, affording ketones 71 in 62–75% yields (Scheme 18, eqn (3)). When substrates 72 bearing two methyl substituents reacted under the standard reaction conditions, the products 73 were formed after 1,2-alkyl migration in good yields (Scheme 18, eqn (4)).
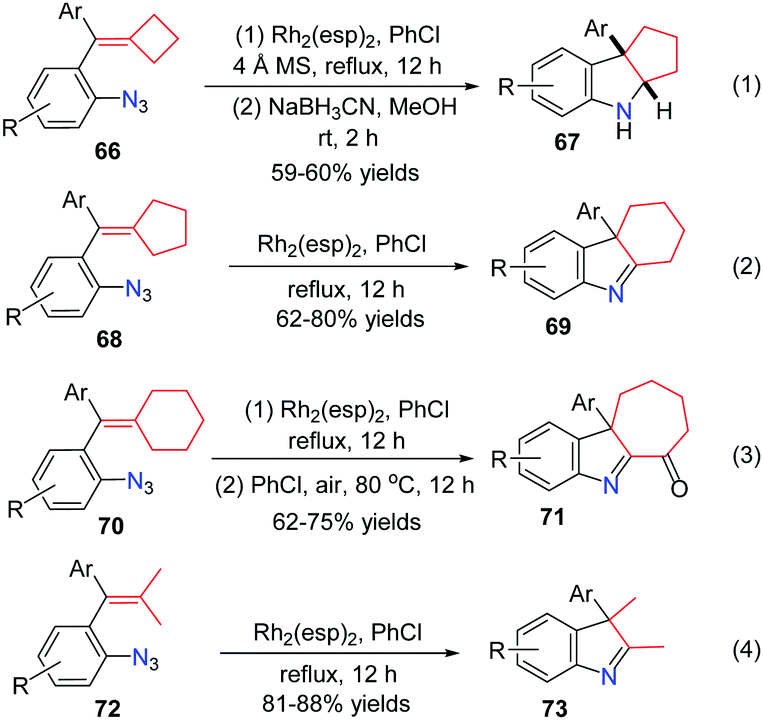 | ||
| Scheme 18 Further substrate scope study. | ||
A plausible pathway accounting for these reactions is outlined in Scheme 19. Without the radical clock in this reaction, ring-opening of intermediate 64 cannot take place and it undergoes another SET to give spirocyclic cationic intermediate 74. A consecutive 1,2-alkyl shift occurs to afford the fused indole 75, which produces the corresponding 3H-indole products.
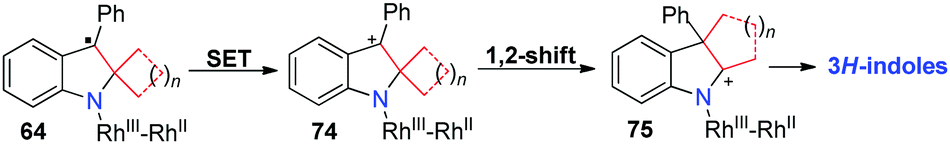 | ||
| Scheme 19 A plausible mechanism for the formation of 3H-indoles. | ||
The active intermediate 62 also attracted our attention strongly and the multicomponent tandem reaction of Rh-nitrenes with ACPs should be very attractive because the incorporation of a nitrogen atom can potentially give rise to novel N-containing heterocycles. Furthermore, extended application to intercept the in situ generated rhodium nitrene to embed the 3C synthon of MCPs into other ring systems, especially heterocycles instead of the indole-fused azetidine 61 formation, seems to be challenging and interesting. As a result, we found that azide-MCPs 60 could undergo intermolecular cyclization with isonitriles catalyzed by the RhII catalyst to produce a series of pyrrole-fused quinolines 76 in good yields via carbodiimide intermediates 78 (Scheme 20).18
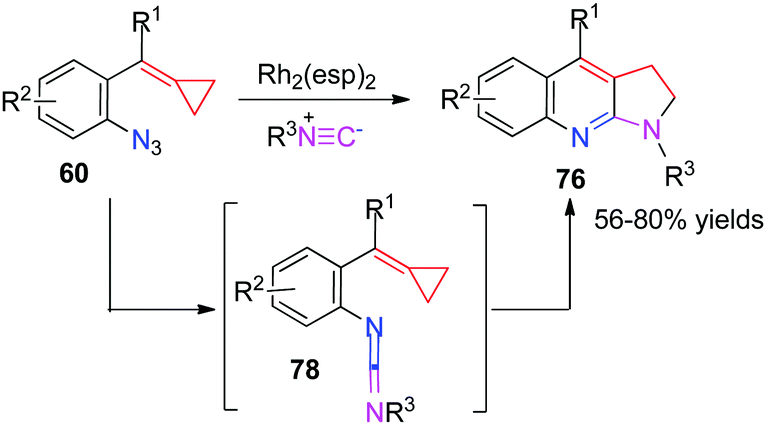 | ||
| Scheme 20 RhII-Catalyzed intermolecular cyclization with isonitriles of azide-ACPs 60. | ||
Two plausible pathways accounting for this intermolecular cascade reaction are outlined in Scheme 21. In path A, the denitrogenation process gives Rh-nitrene 62, which reacts with tBuNC to afford the key intermediate 77. Alternatively (path B), the reaction of Rh2(esp)2 with tBuNC gives Rh2(esp)2(CNtBu)2, which is not very stable and decomposes into Rh2(esp)2(CNtBu) at the same time. Upon regeneration of the catalyst, key intermediate 77 can also be afforded from azide-ACP 60. After the regeneration of the RhII catalyst, 77 is transformed to carbodiimide 78. A consecutive 6π-electrocyclization occurs to afford intermediate 79, which subsequently undergoes a thermal-induced rearrangement to produce pyrrole-fused quinolines 76.
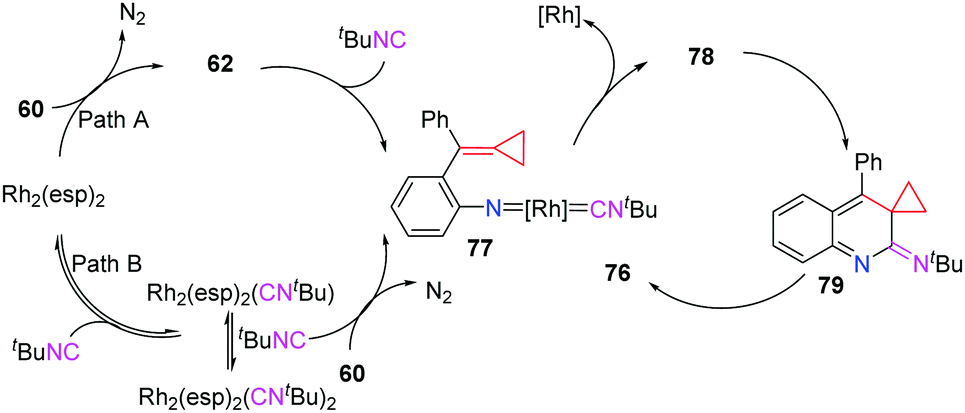 | ||
| Scheme 21 A plausible mechanism for this reaction. | ||
Next, we carried out some transformations of products 76 to improve the applicability of this methodology. For example, after removal of the PMB of product 76a and further condensation with 3,4,5-trimethoxybenzoic acid, DU-145 cell inhibitor 80 could be obtained in 80% yield. It showed the best IC50 value (0.114 μM) and may be used as a novel anti-cancer agent in prostate cancer therapy (Scheme 22).
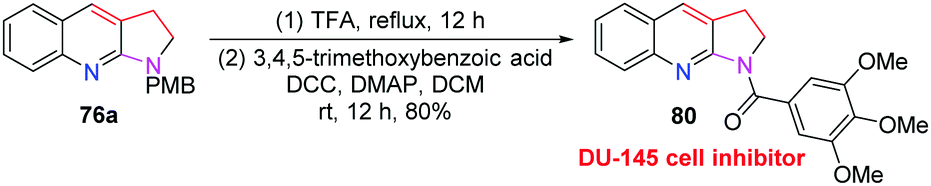 | ||
| Scheme 22 Transformation of compound 76a to 80. | ||
2.2 Transformations of alkyl-FACPs
ACPs and MCPs have proven to be versatile three-carbon synthons for an array of metal-catalyzed annulation reactions. Mascareñas and López reported that FACPs could be used in Pd-catalyzed [3+2+2] cycloadditions with activated alkenes involving ACP proximal bond cleavage.19 The same group also reported Ni-catalyzed intramolecular [3+2+2] cycloaddition with similar substrates.20 It should be noted that Evans and colleagues reported the first example of intermolecular RhI-catalyzed [3+2+2] carbocyclization of FACPs 81 with activated alkynes 82 to get bicycloheptadienes 83 and 84 in good yields in 2008 (Scheme 23).21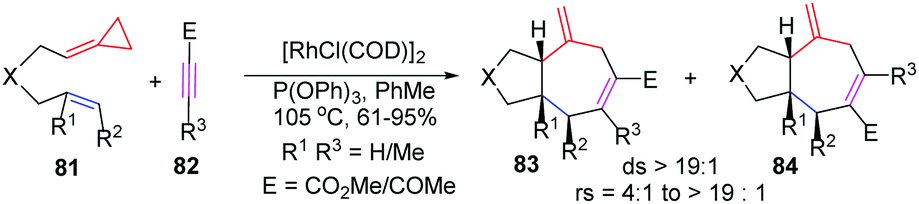 | ||
| Scheme 23 RhI-Catalyzed [3+2+2] carbocyclization of FACPs 81 with activated alkynes 82. | ||
In 2014, López and Mascareñas achieved the RhI-catalyzed [3+2+2] cycloaddition of FACPs 85 with intramolecular alkenes and alkynes. The transformation afforded synthetically relevant 5,7,5-fused tricyclic systems 86 and/or 87 with moderate or good yields, good versatility, and high diastereoselectivities (Scheme 24).22 In the same year, they also reported highly diastereo- and chemoselective Ni0-catalyzed intramolecular [3+2+2] cycloaddition of similar FACPs 85. The reaction proceeded via proximal cleavage of cyclopropane and made it possible to construct relevant 6,7,5-tricyclic frameworks 88 in a one-pot reaction manner. They also pointed out that the characteristics of the nickel ligands were important to the reaction outcome (Scheme 25).23
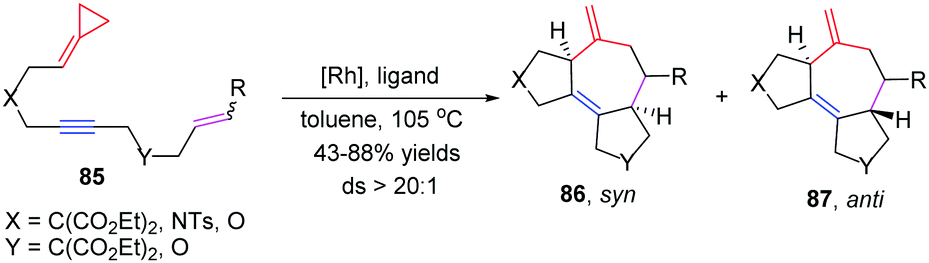 | ||
| Scheme 24 RhI-Catalyzed [3+2+2] cycloaddition of FACPs 85. | ||
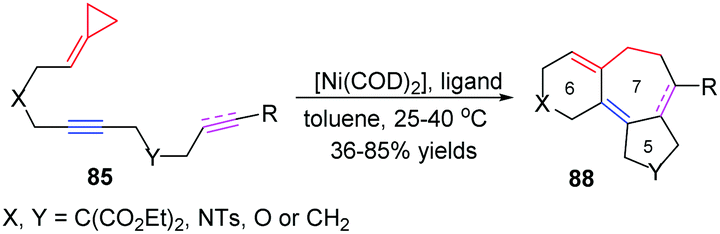 | ||
| Scheme 25 Ni0-Catalyzed [3+2+2] cycloaddition of FACPs 85. | ||
In 2012, Evans's group reported a novel RhI-catalyzed [3+2+1] carbocyclization of carbo- and heteroatom-tethered FACPs 81 with CO to realize the stereoselective construction of cis-fused bicyclohexenones 89 in good yields (Scheme 26).24
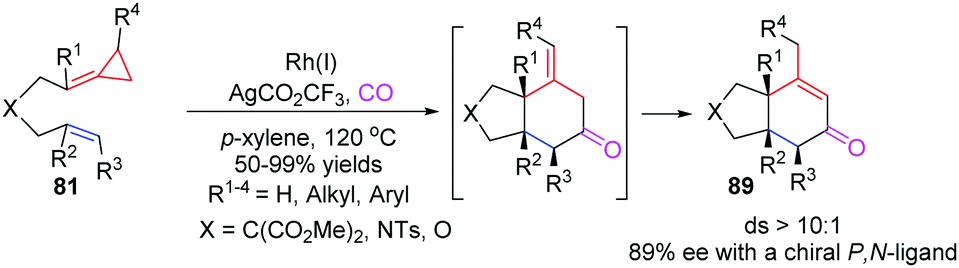 | ||
| Scheme 26 RhI-Catalyzed [3+2+1] carbocyclization of FACPs 81 with CO. | ||
Thereafter, the development of the RhI-catalyzed [(3+2)+1] carbocyclization reaction of alkynylidenecyclopropanes 81 with carbon monoxide to construct polysubstituted phenols 90 is described by the groups of Evans and Chung, respectively (Scheme 27).25 This work offers a convenient method for the selective formation of 5,6-bicyclic phenols, which provide important intermediates for the target directed synthesis of natural products such as benfluron and MN100.
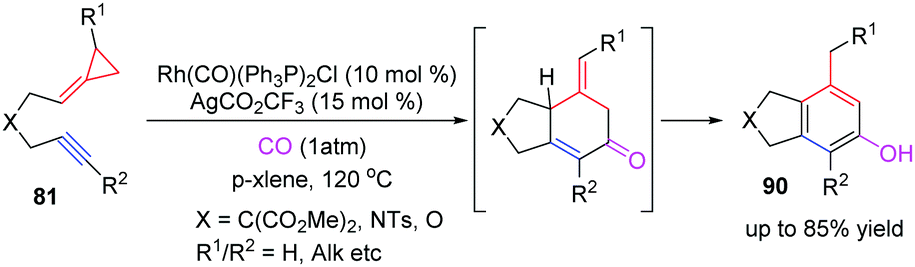 | ||
| Scheme 27 RhI-Catalyzed [(3+2)+1] carbocyclization reaction of FACPs 81 with CO. | ||
In 2012, in the absence of reaction partners, such as activated alkynes and carbon monoxide, they reported the novel diastereoselective RhI-catalyzed ene-cycloisomerization (ECI) reactions of alkenylidenecyclopropanes 81 to provide an atom-economical approach to five-membered carbo- and heterocycles 91 that contain two new stereogenic centers in good yields (Scheme 28).26
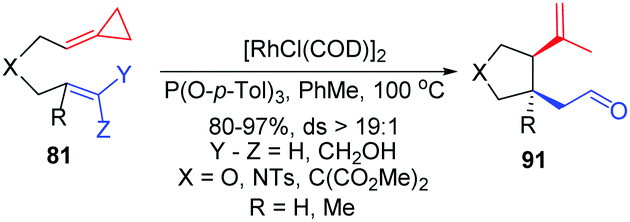 | ||
| Scheme 28 RhI-Catalyzed ene-cycloisomerization (ECI) reactions of FACPs 81. | ||
More importantly, the isolation and characterization of a novel rhodacycle intermediate 92 implicated in the Rhl-catalyzed reactions of FACPs was achieved by Evans's group (Scheme 29).27 The structure of the metallacycle was unambiguously determined by X-ray crystallography. The metallacycle 92 is catalytically competent in RhI-catalyzed carbocyclization with alkynes and carbon monoxide along with RhI-catalyzed ene-cycloisomerization (ECI) in the absence of a π-component under standard conditions. These reactions proceeded through the RhIII metallacycle. They isolated the metallacycle and provided important insight into the ligand requirements for the insertion of π components. Furthermore, they also described a novel [(3+2)+2] carbocyclization reaction of the metallacycle 92 with an activated allene to produce 93.
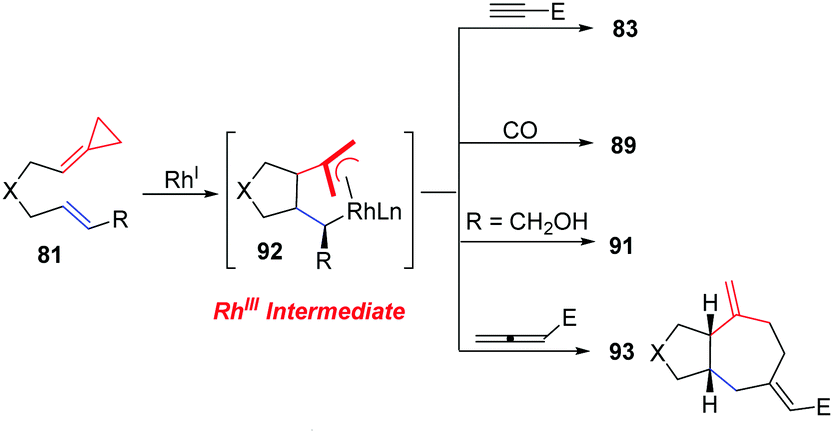 | ||
| Scheme 29 Stereoselective RhI-catalyzed [(m+n)+o] carbocyclizations and ene-cycloisomerizations (ECI) of FACPs 81via intermediates 92. | ||
In 2013, our group developed a new RhI-catalyzed intramolecular cycloisomerization reaction of nitrogen-tethered indoles with FACPs 94 to provide easy access to tetrahydro-β-carboline derivatives 95 in good yields (Scheme 30).28 The reaction mechanism is proposed on the basis of isotopic labeling and control experiments, and is also supported by DFT calculations.
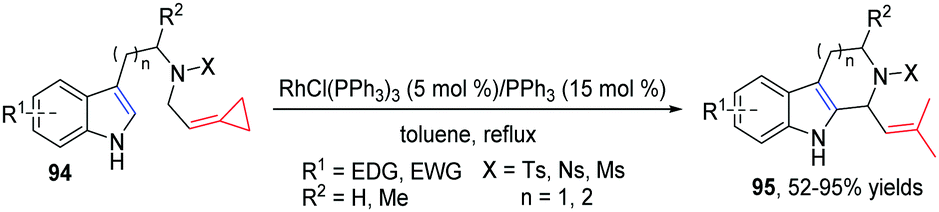 | ||
| Scheme 30 RhI-Catalyzed cycloisomerization reaction of FACPs 94. | ||
A plausible mechanism for this reaction is outlined in Scheme 31. Initial insertion of the metal at the distal position of alkylidenecyclopropane 94 gives metallacyclobutene 96, followed by isomerization to form intermediate 97 through a trimethylenemethane (TMM)-like transition state. Intermediate 97 undergoes β-H elimination to give Rh–H species 98, which can isomerize to produce π-allylic RhIII complex 99. Conjugated diene 100 can be obtained from intermediate 99 through reductive elimination along with regeneration of the RhI complex. There are two competing pathways for cyclization in the Pictet–Spengler reaction, since both the indole C2- and C3-positions are nucleophilic. Through an indole C3-position attack on the conjugate diene, spiroindolenine intermediate 101 is formed, which can further undergo a 1,2-alkyl shift to afford the six-membered-ring intermediate 102. Alternatively, nucleophilic attack of the indole C2-position on the diene moiety leads directly to intermediate 102, followed by deprotonation to give 95.
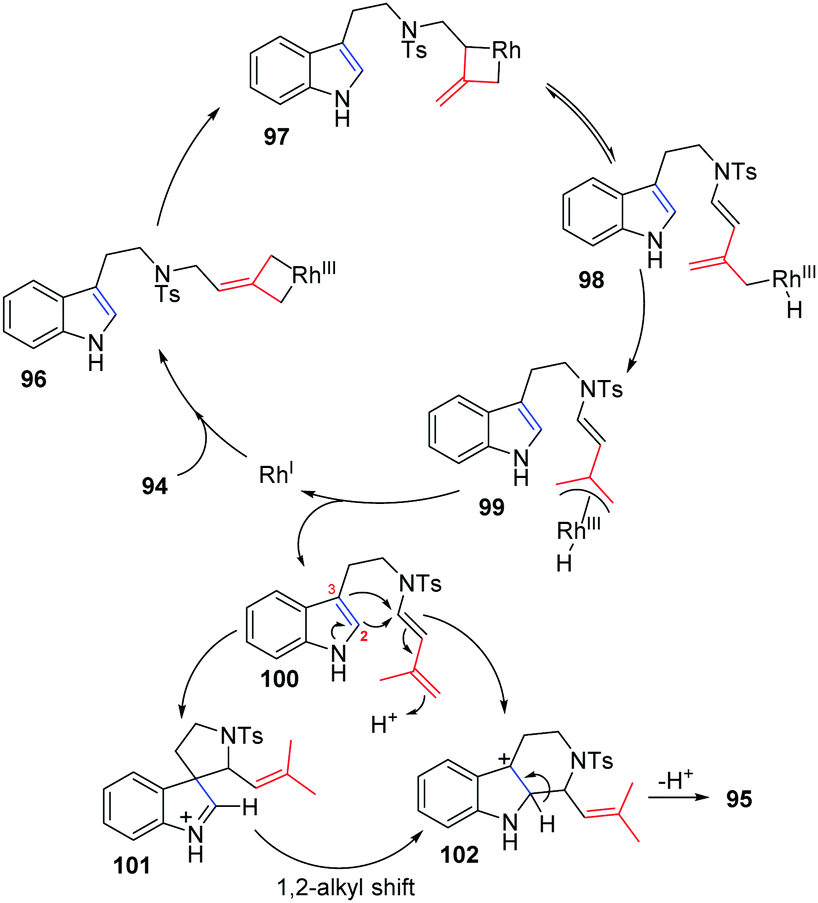 | ||
| Scheme 31 A plausible mechanism for this reaction. | ||
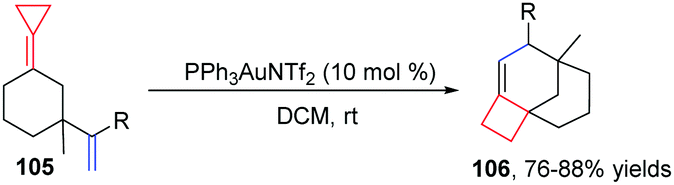
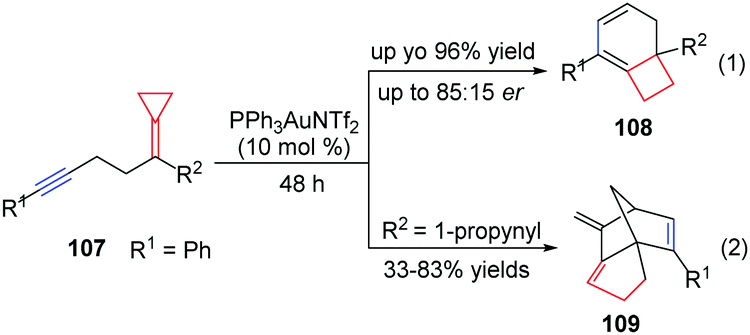
Taking advantage of the ring strain relief strategy, Gagné's group recently reported several AuI-catalyzed rearrangements of FACPs. In 2012, they found the first enantioselective AuI-catalyzed Cope rearrangement from achiral 1,5-dienes 103 to form products 104 (Scheme 32).29 Subsequently, they investigated ACP-containing cyclic 1,5-dienes 105 and disclosed an AuI-catalyzed ring-expanding cycloisomerization of 105 to produce the corresponding bicyclo[4.2.0]oct-1-enes 106 in good yields (Scheme 33).30 Based on this result, novel ACP-bearing 1,5-enynes such as 107 were synthesized, and under the catalysis of PPh3AuNTf2, bicyclo[4.2.0]dienes 108 were obtained through a sequential 6-endo-dig-cyclization/ring expansion/net 1,2-hydrogen shift process (Scheme 34, eqn (1)).31 It is worth noting that when FACPs 107 (R2 = 1-propynyl) were treated with PPh3AuNTf2 (10 mol%) in 1,2-dichloroethane at 50 °C, the expected bicyclic products 108 were not formed. Instead, the tricyclic compounds 109 were obtained as a single diastereomer in 33–83% yields (Scheme 34, eqn (2)).32
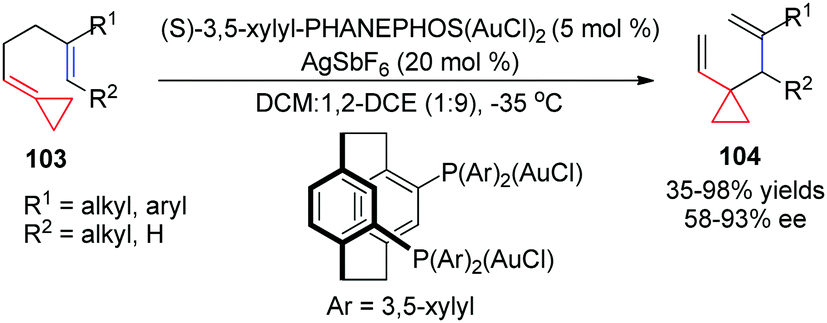 | ||
| Scheme 32 AuI-Catalyzed Cope rearrangement of FACPs 103. | ||
 | ||
| Scheme 33 AuI-Catalyzed ring-expanding cycloisomerization of FACPs 105. | ||
 | ||
| Scheme 34 AuI-Catalyzed rearrangements of FACPs 107. | ||
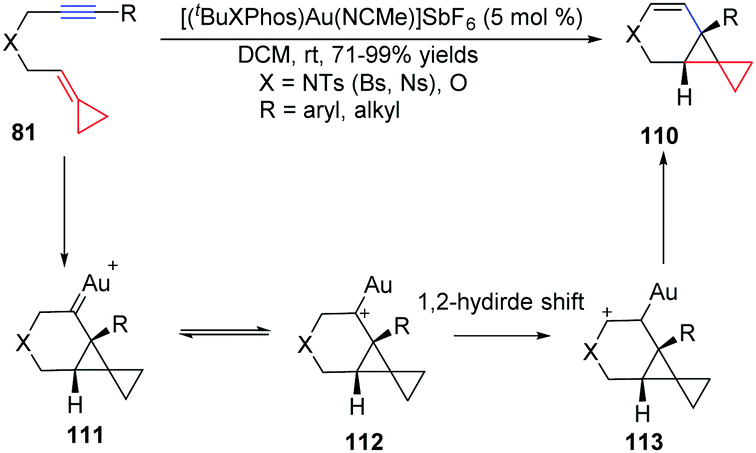
Recently, we have also developed another example of AuI-catalyzed cycloisomerization of nitrogen and oxygen-tethered alkylidenecyclopropanes 81 to provide easy access to tricyclic compounds or bicyclo[4.1.0]heptene derivatives 110 in high yields under very mild conditions (Scheme 35).33 The reaction may initially form cyclopropyl gold–carbene intermediate 111, which equilibrates with intermediate 112, followed by a 1,2-hydride shift process.
 | ||
| Scheme 35 AuI-Catalyzed cycloisomerization of FACPs 81. | ||
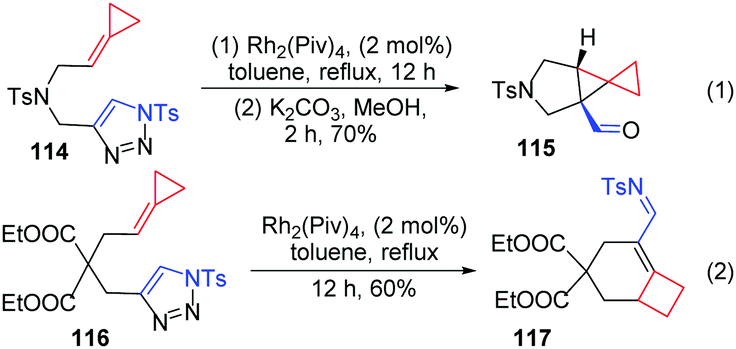
Moreover, our group also prepared triazole-tethered alkyl-FACPs 114 and 116, and azavinyl carbenes also reacted smoothly with alkylidenecyclopropane to furnish two structurally valuable products, 115 and 117.16 Tricyclic aldehyde 115 was formed in 70% yield after cyclopropanation and hydrolysis when N-tethered-triazole-ACP 114 was used as a substrate (Scheme 36, eqn (1)). Interestingly, the reaction of substrate 116 delivered trans-α,β-unsaturated imine product 117 in 60% yield (Scheme 36, eqn (2)).
 | ||
| Scheme 36 Rh-Catalyzed triazole-tethered FACPs 114 and 116. | ||
2.3 Transformations of ring-FACPs
When functional groups are introduced at the ring of MCPs or ACPs, the ring-opening or ring-expansion of FACPs may take place in a very special and different manner. In 2011, our group reported a TiCl4-mediated intramolecular ring enlargement of FACPs 118 and 119 with propargylic esters to afford the corresponding chlorinated bicyclo[4.2.0]oct-5-ene derivatives 120 in moderate to good yields (Scheme 37).34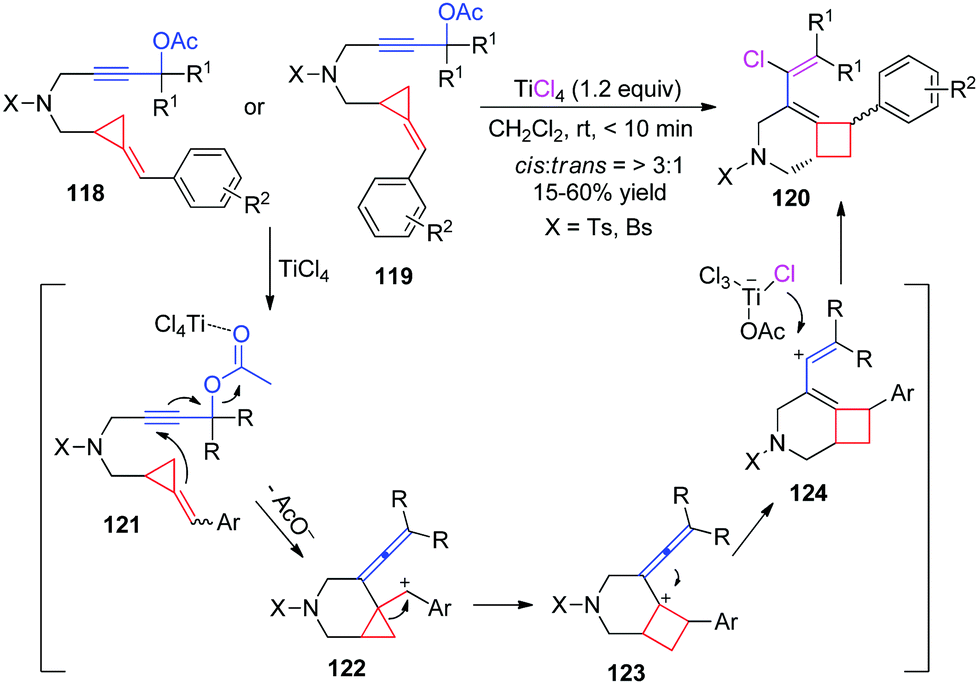 | ||
| Scheme 37 TiCl4-Mediated intramolecular ring enlargement of FACPs 118 or 119. | ||
We proposed a plausible reaction mechanism for this TiCl4-mediated carbocyclization in Scheme 38. Coordination of the ester group to TiCl4 gives intermediate 121. Nucleophilic intramolecular addition of the pendant methylenecyclopropane to the alkyne moiety along with the release of an acyloxy group affords carbocation 122, which contains a vinylidene moiety. Subsequently, carbocationic intermediate 122 undergoes intramolecular ring enlargement of cyclopropane via 1,2-migration giving intermediate 123 which can give vinyl cationic intermediate 124 through isomerization. Then, a chloride ion is transferred to a vinyl cation from the in situ generated metal complex, affording the corresponding chlorinated bicyclo[4.2.0]oct-5-ene 120. It should be noted that carbocation 122 can be stabilized by the neighboring aryl unit, which can serve as a driving force for this transformation. Moreover, as can be seen from Scheme 38, E- and Z-methylenecyclopropanes 118 or 119 give the same benzylic cation 122 under identical conditions. This can explain why the same products could be formed using 118 and 119 as the substrates.
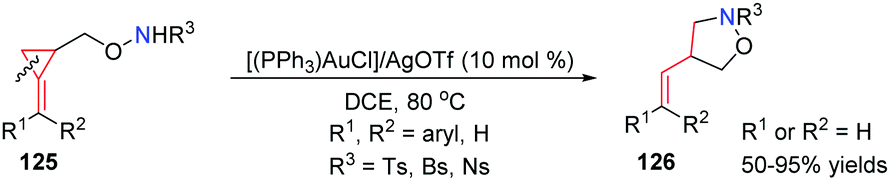 | ||
| Scheme 38 AuI-Catalyzed reaction of FACPs 125. | ||
In 2012, our group reported an interesting and novel AuI-catalyzed intramolecular hydroamination and ring-opening of sulfonamide-substituted 2-(arylmethylene)cyclopropylcarbinols 125. A variety of 4-substituted isoxazolidine derivatives 126 were obtained in good to high yields through highly regioselective cleavage of a C–C single bond in the cyclopropane unit (Scheme 38).35
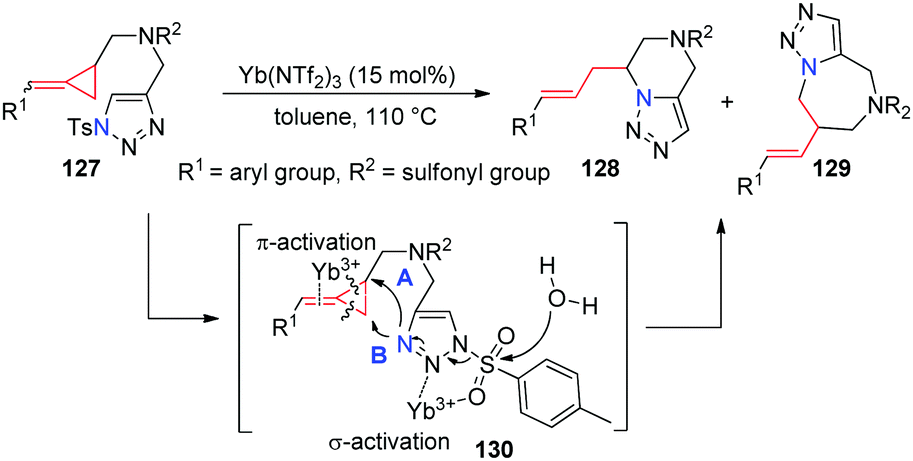
Recently, our group successfully prepared a series of novel methylenecyclopropane-triazoles 127. Intramolecular nucelophilic cyclizations of them have been explored in the presence of a Yb(NTf2)3 catalyst, giving the corresponding triazole containing six- and seven-membered heterocycles 128 and 129 in good yields upon heating in toluene (Scheme 39).36 In this reaction, Lewis acid Yb(NTf2)3 first coordinates to the Ts-triazole moiety through σ-activation and the alkene moiety in MCP parts via π-activation to give intermediate 130, which may then undergo intramolecular cyclization via two possible pathways along with the hydrolysis of a sulfonyl group by ambient water to give the corresponding products 128 (path A) and 129 (path B).
 | ||
| Scheme 39 Yb(NTf2)3 catalyzed intramolecular nucelophilic cyclizations of FACPs 127. | ||
3. Conclusion and outlook
In summary, the development of FACP chemistry has attracted widespread attention from chemists. Different types of FACP, which can be easily prepared, emerge as endless useful substrates in organic synthesis. The reaction products of FACPs have important structure motifs in organic and medicinal chemistry. These new findings subsequently enrich the strained small carbocyclic chemistry at the present stage. Furthermore, these novel synthetic methodologies have some potential applications in the synthesis of natural products and bioactive compounds. As a result, designing and discovering more novel types of FACP and using them to synthesize targeted natural products is gradually becoming the tendency in the field of FACP chemistry. Furthermore, investigating the reactivity of heterocyclic FACPs and realizing the C–H bond activation instead of C–C bond activation of some special FACPs will be more challenging and interesting (Scheme 40). | ||
| Scheme 40 Tendency and challenge of the development of FACPs. | ||
Acknowledgements
We are grateful for financial support from the National Basic Research Program of China (973)-2015CB856603, the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB20000000, and the National Natural Science Foundation of China (20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, and 20732008).Notes and references
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef CAS PubMed; (b) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed; (d) L.-X. Shao and M. Shi, Curr. Org. Chem., 2007, 11, 1135 CrossRef CAS; (e) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed; (f) H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS; (g) G. Audran and H. Pellissier, Adv. Synth. Catal., 2010, 352, 575 CrossRef CAS; (h) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (i) L. Yu and R. Guo, Org. Prep. Proc. Int., 2011, 43, 209 CrossRef CAS; (j) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641 CrossRef CAS PubMed; (k) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (l) H. Pellissier, Tetrahedron, 2014, 70, 4991 CrossRef CAS; (m) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS PubMed; (n) L. Yu, M. Liu, F. Chen and Q. Xu, Org. Biomol. Chem., 2015, 13, 8379 RSC.
- For selected examples, see: (a) N. Tsukada, A. Shibuya, I. Nakamura and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 8123 CrossRef CAS; (b) A. I. Siriwardana, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2004, 69, 320 CrossRef PubMed; (c) A. Fürstner and C. Aïssa, J. Am. Chem. Soc., 2006, 128, 6306 CrossRef PubMed; (d) M. Shi, L.-P. Liu and J. Tang, J. Am. Chem. Soc., 2006, 128, 7430 CrossRef CAS PubMed; (e) C. Aïssa and A. Fürstner, J. Am. Chem. Soc., 2007, 129, 14836 CrossRef PubMed; (f) S. Simaan and I. Marek, J. Am. Chem. Soc., 2010, 132, 4066 CrossRef CAS PubMed; (g) K. Chen, M. Jiang, Z. Zhang, Y. Wei and M. Shi, Eur. J. Org. Chem., 2011, 7189 CrossRef CAS; (h) S. Cui, Y. Zhang and Q. Wu, Chem. Sci., 2013, 4, 3421 RSC; (i) J. C. Timmerman, B. D. Robertson and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015, 54, 2251 CrossRef CAS PubMed; (j) Z.-Z. Zhu, K. Chen, L.-Z. Yu, X.-Y. Tang and M. Shi, Org. Lett., 2015, 17, 5994 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Shi and B. Xu, Org. Lett., 2002, 4, 2145 CrossRef CAS PubMed; (b) M. Shi, B. Xu and J.-H. Huang, Org. Lett., 2004, 6, 1175 CrossRef CAS PubMed; (c) L.-X. Shao, B. Xu, J.-W. Huang and M. Shi, Chem. – Eur. J., 2006, 12, 510 CrossRef PubMed; (d) M. E. Scott, Y. Bethuel and M. Lautens, J. Am. Chem. Soc., 2007, 129, 1482 CrossRef CAS PubMed; (e) L. Yu, J. Meng, L. Xia and R. Guo, J. Org. Chem., 2009, 74, 5087 CrossRef CAS PubMed.
- For selected examples, see: (a) B. Xu, Y. Chen and M. Shi, Tetrahedron Lett., 2002, 43, 2781 CrossRef CAS; (b) L.-P. Liu and M. Shi, Chem. Commun., 2004, 2878 RSC; (c) J.-W. Huang and M. Shi, J. Org. Chem., 2005, 70, 3859 CrossRef CAS PubMed; (d) T. Kippo, K. Hamaoka and I. Ryu, J. Am. Chem. Soc., 2013, 135, 632 CrossRef CAS PubMed; (e) L. Yu, Y. Wu, T. Chen, Y. Pan and Q. Xu, Org. Lett., 2013, 15, 144 CrossRef CAS PubMed.
- For selected examples, see: (a) I. Nakamura, T. Nemoto, Y. Yamamoto and A. de Meijere, Angew. Chem., Int. Ed., 2006, 45, 5176 CrossRef CAS PubMed; (b) I. Nakamura, R. Nagata, T. Nemoto, M. Terada, Y. Yamamoto, T. Späth and A. de Meijere, Eur. J. Org. Chem., 2007, 4479 CrossRef CAS; (c) X.-Y. Tang and M. Shi, J. Org. Chem., 2009, 74, 5983 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Liang, L. Jiao, Y. Wang, Y. Chen, L. Ma, J. Xu, S. Zhang and Z.-X. Yu, Org. Lett., 2006, 8, 5877 CrossRef CAS PubMed; (b) W. Li and M. Shi, Tetrahedron, 2007, 63, 11016 CrossRef CAS; (c) D.-H. Zhang, Y. Wei and M. Shi, Eur. J. Org. Chem., 2011, 4940 CAS; (d) R. Sang, X.-Y. Tang and M. Shi, Org. Chem. Front., 2014, 1, 770 RSC.
- A. I. Siriwardana, M. Kamada, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2005, 70, 5932 CrossRef CAS PubMed.
- K. Chen, Z. Zhang, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 7696 RSC.
- L.-Z. Yu, X.-B. Hu, Q. Xu and M. Shi, Chem. Commun., 2016, 52, 2701 RSC.
- K. Chen, R. Sun, Q. Xu, Y. Wei and M. Shi, Org. Biomol. Chem., 2013, 11, 3949 CAS.
- L.-Z. Yu, Q. Xu, X.-Y. Tang and M. Shi, ACS Catal., 2016, 6, 526 CrossRef CAS.
- B. Yao, Y. Li, Z. Liang and Y. Zhang, Org. Lett., 2011, 13, 640 CrossRef CAS PubMed.
- S. Li, Y. Luo and J. Wu, Org. Lett., 2011, 13, 3190 CrossRef CAS PubMed.
- S. Li, Z. Li and J. Wu, Adv. Synth. Catal., 2012, 354, 3087 CrossRef CAS.
- K. Chen, Z.-Z. Zhu, Y.-S. Zhang, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 6645 CrossRef CAS PubMed.
- K. Chen, Z.-Z. Zhu, J.-X. Liu, X.-Y. Tang, Y. Wei and M. Shi, Chem. Commun., 2016, 52, 350 RSC.
- K. Chen, X.-Y. Tang and M. Shi, Chem. Commun., 2016, 52, 1967 RSC.
- (a) A. Delgado, J. R. Rodriguez, L. Castedo and J. L. Mascareñas, J. Am. Chem. Soc., 2003, 125, 9282 CrossRef CAS PubMed; (b) J. Duran, M. Gulísa, L. Casteso and J. L. Mascareñas, Org. Lett., 2005, 7, 5693 CrossRef CAS PubMed.
- L. Saya, G. Bhargava, M. A. Navarro, M. Gulias, F. López, I. Fernandez, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2010, 49, 9886 CrossRef CAS PubMed.
- P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2008, 130, 12838 CrossRef CAS PubMed.
- M. Araya, M. Gulías, I. Fernández, G. Bhargava, L. Castedo, J. L. Mascareñas and F. López, Chem. – Eur. J., 2014, 20, 10255 CrossRef CAS PubMed.
- L. Saya, I. Fernández, F. López and J. L. Mascareñas, Org. Lett., 2014, 16, 5008 CrossRef CAS PubMed.
- S. Mazumder, D. Shang, D. E. Negru, M.-H. Paik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569 CrossRef CAS PubMed.
- (a) P. A. Evans, A. J. Burnie and D. E. Negru, Org. Lett., 2014, 16, 4356 CrossRef CAS PubMed; (b) S. Kim and Y. K. Chung, Org. Lett., 2014, 16, 4352 CrossRef CAS PubMed.
- P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2012, 134, 3635 CrossRef CAS PubMed.
- P. A. Inglesby, J. Bacsa, D. E. Negru and P. A. Evans, Angew. Chem., Int. Ed., 2014, 53, 3952 CrossRef CAS PubMed.
- D.-H. Zhang, X.-Y. Tang, Y. Wei and M. Shi, Chem. – Eur. J., 2013, 19, 13668 CrossRef CAS PubMed.
- R. J. Felix, D. Weber, O. Gutierrez, D. J. Tantillo and M. R. Gagné, Nat. Chem., 2012, 4, 405 CrossRef CAS PubMed.
- R. J. Felix, O. Gutierrez, D. J. Tantillo and M. R. Gagné, J. Org. Chem., 2013, 78, 5685 CrossRef CAS PubMed.
- H. Zheng, R. J. Felix and M. R. Gagné, Org. Lett., 2014, 16, 2272 CrossRef CAS PubMed.
- H. Zheng, L. L. Adduci, R. J. Felix and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 7904 CrossRef CAS PubMed.
- D.-H. Zhang, Y. Wei and M. Shi, Chem. – Eur. J., 2012, 18, 7026 CrossRef CAS PubMed.
- Z. Zhang and M. Shi, Tetrahedron Lett., 2011, 52, 6541 CrossRef CAS.
- D.-H. Zhang, K. Du and M. Shi, Org. Biomol. Chem., 2012, 10, 3763 CAS.
- R. Sun, D.-H. Zhang and M. Shi, Synlett, 2014, 2293 CAS.
| This journal is © The Royal Society of Chemistry 2017 |