
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A Co-based single-molecule magnet confined in a barium phosphate apatite matrix with a high energy barrier for magnetization relaxation†
Pavel E.
Kazin
 *a,
Mikhail A.
Zykin
b,
Lev A.
Trusov
b,
Artem A.
Eliseev
b,
Oxana V.
Magdysyuk
c,
Robert E.
Dinnebier
d,
Reinhard K.
Kremer
d,
Claudia
Felser
e and
Martin
Jansen
de
*a,
Mikhail A.
Zykin
b,
Lev A.
Trusov
b,
Artem A.
Eliseev
b,
Oxana V.
Magdysyuk
c,
Robert E.
Dinnebier
d,
Reinhard K.
Kremer
d,
Claudia
Felser
e and
Martin
Jansen
de
aDepartment of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia. E-mail: kazin@inorg.chem.msu.ru; Tel: + 7 495 9393440
bDepartment of Materials Science, Lomonosov Moscow State University, 119991 Moscow, Russia
cDiamond Light Source Ltd, Harwell Science and Innovation Campus, OX11 0DE Didcot, UK
dMax Planck Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany
eMax Planck Institute for Chemical Physics of Solids, Nöthnitzer Str. 40, 01187 Dresden, Germany
First published on 20th April 2017
Abstract
An apatite-type barium phosphate with a high content of cobalt ions in the trigonal channels features slow relaxation of magnetization with an energy barrier Ueff of up to 387 cm−1, which is well above the values for all so far known d-metal based single-molecule magnets (SMMs).
Consisting of an atomic group with only one paramagnetic atom in a highly anisotropic environment, a mononuclear SMM also named a single-ion magnet (SIM) may perform as a molecule-size permanent magnet which reveals quantum behavior and may serve as a tiniest element in ultrahigh-density magnetic memory, molecular electronic, and spintronic devices.1 In order to enable its application, materials exhibiting higher blocking temperatures (Tb) and hence higher energy barriers (Ueff) for magnetization reversal are required. These features are connected to huge single-ion magnetic anisotropy arising from an efficient spin–orbital coupling (SOC) acting in an anisotropic (axial) crystal field.2 So far, the highest Ueff values exceeding 1000 cm−1 have been encountered in complexes of rare-earth elements, however Tb did not scale up accordingly due to non-thermal relaxation pathways.3,4 Compounds of d-elements commonly exhibit lower Ueff values: among the largest are 246 cm−1 for a linear coordinated Fe1+ methanide5 and 180 cm−1 for a Co2+ clathrochelate,6 both representing Kramers' ions with a d7 configuration. Recent magnetization studies on Li3−xFexN have suggested that the linear coordinated Fe1+ ion also behaves like a SIM with Ueff = 300 cm−1.7 Co2+ is of special interest as it is common in stable compounds and may keep a large orbital moment in a crystal field so that a maximal value of magnetic anisotropy is achieved.8 Recently, we have found that small magnetically anisotropic atomic groups – dioxometallate anions – inserted into the channels of an apatite lattice revealed SIM features: the paramagnetic linear [OCuO]−-anions show Ueff values of up to ca. 100 cm−1,9,10 while for the bent [OCoO]2−-anions values of 51–59 cm−1 have been recorded.11 Remarkably, at a low Co concentration, a part of the magnetization exhibits a new very slow relaxation (VSR) process with an unusually high Ueff of 200–275 cm−1. The precise nature of that remained unclear though. Since the most promising barium compound contained only 0.04 mol Co per formula unit and only 30% of its magnetization relaxed that way, we have made further efforts in optimizing these numbers. Indeed, we have managed to insert a large quantity of Co ions into the channels of the apatite-type barium phosphate. Here we present the magnetic properties of the new materials obtained, demonstrating VSR of the major part of the magnetization with Ueff values considerably exceeding all those found before for open d-shell transition elements.
Samples with nominal composition Ba10(PO4)6(CoxOH1−2x)2, where x = 0.05 (1), 0.3 (2, 2a), and 0.5 (3), were prepared as described in the ESI.† The final annealing was conducted at 1450 °C for 1 and 2 and 1400 °C for 2a and 3. According to XRD the samples represent an apatite phase sometimes with a small admixture of Ba3(PO4)2 and CoO (Fig. S1–S4, ESI†). SEM/EDX of 3 reveals a polycrystalline barium phosphate apatite matrix containing Co in a quantity corresponding to x ≈ 0.3, and CoO as micron-sized rounded grains embedded.
The crystal structure details of 3 were determined by Rietveld refinement of the synchrotron powder XRD pattern (the Diamond Light Source, United Kingdom, Fig. S1 and Tables S1–S3, ESI†).‡ The compound crystallizes in the apatite structure, space group P63/m, a = 10.1926(1), c = 7.7532(1) Å. A Co-atom is located in the trigonal channel with the refined site occupancy corresponding to x = 0.33, and is randomly displaced from the ideal (0,0,0) position within the ab-plane (Fig. 1). Due to the two short contacts of Co with intra-channel oxygen atoms, a bent [OCoO] atomic group results, resembling the one found in the Co-doped strontium apatite.11,12 Since further Co–O distances are greater than 3 Å, Co displays a coordination number of two. When the [OCoO] anion represents a separate unit in the crystal lattice, the possible Co–Co interatomic distances cannot be markedly below the values of the cell parameters. This suggests considering [OCoO] as a paramagnet which is well diluted in a diamagnetic matrix and is not involved in appreciable exchange and dipole interactions. The total content of Co in the sample, estimated from the XRD quantities of the apatite and CoO phases, corresponds to x = 0.47 which is close to the nominal x = 0.5. This confirms that the major amount of Co diluted in the apatite phase is found in the trigonal channels. All other samples were analyzed using XRD (Bruker D8 diffractometer, CuKα1 radiation). The powder XRD patterns of 2 and 2a were refined with somewhat lower accuracy, though still allowing us to localize a Co-atom near (0,0,0) with the occupancy corresponding to x = 0.23 (Fig. S2, S3 and Tables S4, S5, ESI†). A small amount of Co has not entered into the apatite phase and was detected as a CoO impurity (0.3–0.4 wt%). The powder XRD pattern of 1 represented a pure apatite phase (Fig. S4, ESI†). An observed electron density around (0,0,0) could be attributed to intrachannel Co, but its (low) content was not estimated reliably (Table S6, ESI†).
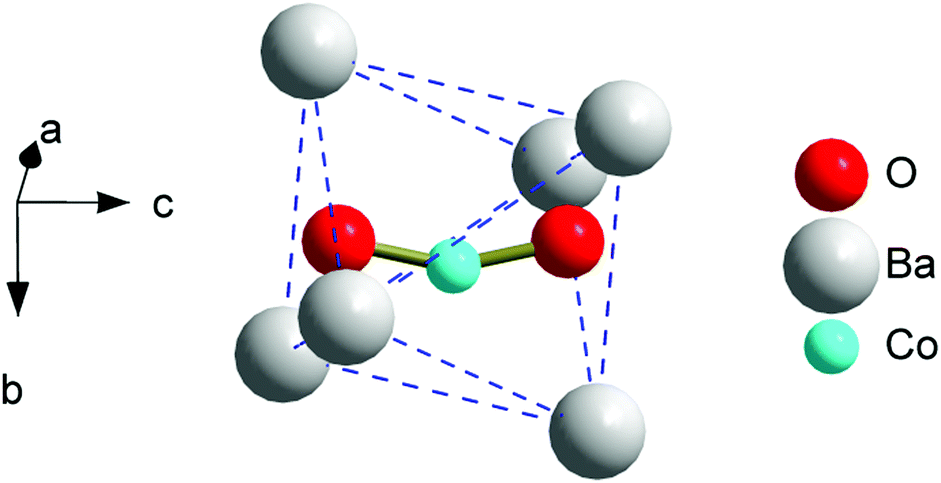 | ||
| Fig. 1 A fragment of the trigonal channel (face-sharing octahedra of Ba atoms extending along the c-axis) in the apatite structure of the as prepared compound with the refined composition of Ba10(PO4)6(Co0.33O0.72)2. | ||
In a previous communication for Co-doped strontium apatites,11 we distinguished three magnetization relaxation regimes contributing to the total magnetization: (i) a field induced “normal” slow relaxation (NSR) with a Ueff of tens of cm−1, covering the major fraction of the magnetization; (ii) a zero field very slow relaxation (VSR) with a Ueff of some two hundreds of cm−1, mostly as a small contribution, and (iii) a paramagnetic behavior of a part of magnetization designated as a fast relaxation (FR). In the Co-doped barium apatites presented here, the frequency dependence of the ac susceptibility χac(f) measured under a zero dc field and above 10 K reveals a large χ′′(T)-peak connected to VSR (Fig. S5–S8, ESI†). By simultaneously fitting χ′(f) and χ′′(f) in the generalized Debye model13 we determined a relaxation time τ, its distribution width α, and a fraction of VSR susceptibility F (ESI†). An adiabatic (high-frequency) susceptibility value was considered as an FR fraction as soon as we did not observe an NSR checking χac(f) in a magnetic field at low temperatures. The VSR fraction covers the major part of the ac susceptibility and at 10 K comprises 68, 89, 77, and 74% for 1, 2, 2a, and 3 respectively. At 10 K, α is relatively large, and amounts to 0.3–0.5. However, above 30 K, α drops below 0.1, implying a very narrow relaxation time distribution width which distinguishes SMMs.
The relaxation time values are depicted as Arrhenius-type plots in Fig. 2. At low temperatures, τ is limited by quantum tunneling and Raman processes.14 At higher temperatures, the dependence becomes steep and approaches a linear one, indicating that a thermally activated Orbach process is predominant. In the whole temperature range, the data can be fitted with the function11,14
τ−1 = τQTM−1 + BT + CTn + τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ueff/kBT), exp(−Ueff/kBT), | (1) |
 | ||
| Fig. 2 Arrhenius plot for the relaxation time τ calculated from χac(f) measured under a zero dc field; symbols – experimental data, lines – linear fits. Inset: An enlarged area at high temperatures. Sample designation: 1 – triangles, 2 – green circles, 2a – red circles, and 3 – crosses. | ||
The field dependence of a magnetization M(H) at 1.8 K for all the samples (Fig. 3 and Fig. S9, ESI†) shows an irreversibility which is relatively pronounced in intermediate fields, considerably weakened near the zero field, and becomes almost reversible in high fields. This behavior is usually explained considering a direct relaxation process strongly prevailing under high fields and a quantum tunneling of magnetization which is substantial in the zero field and is suppressed when a field is applied.1,14 In accordance with the latter, the dc magnetization decay measured for 3 under a field of 1.5 kOe follows an expanded exponential behavior (Fig. 3 inset) with τ ranging from 44 to 3 s in the 2–8 K temperature interval, while under the zero field the decay is too fast to be registered in a dc mode.
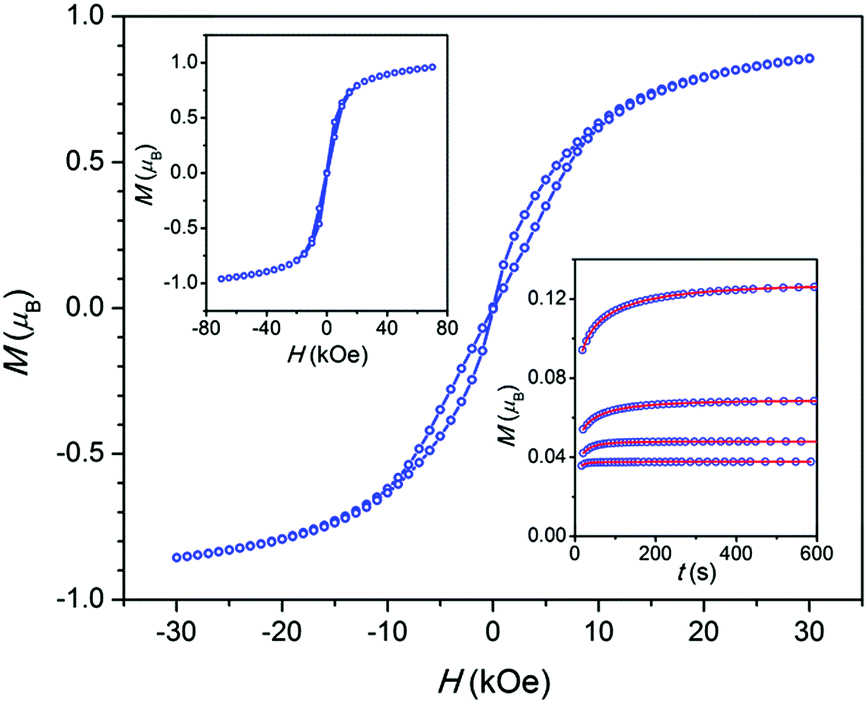 | ||
| Fig. 3 Sample 3. Field dependence of the magnetization per atom of Co at T = 1.8 K measured with a field sweeping rate of 50 Oe s−1. Top-left inset: The same in a larger field interval and at a field sweeping rate of 175 Oe s−1. Lines have been drawn to guide the eye. Bottom-right inset: Magnetization decay after switching on a field of 1.5 kOe at temperatures of 2, 4, 6, and 8 K (curves from top to bottom, respectively), symbols – experimental points, lines – fitting. | ||
The electronic and even oxidation states of a Co-ion in a VSR-center are not yet clear due to the inconclusive results of XANES and red-ox titration.11 In order to clarify these items we will further analyze in detail the dc magnetic properties of 2, which has the highest VSR fraction, taking into consideration the distribution of Co in the sample.
In 2, 74% of the Co resides in the trigonal channels of an apatite phase, ca. 15% as a CoO admixture, and the remainder is not assessed. CoO is an antiferromagnet with TN = 289 K and its χ slightly decreases from 0.005 to 0.0043 emu mol−1 with temperature ranging from TN to 4.2 K.15 Therefore its contribution to the low temperature magnetization is negligibly small (below 0.2%).
First, we have to subtract the FR contribution (11% of the susceptibility at 10 K). For this purpose, we synthesized a sample of barium apatite 4 with a composition different from that of 1 only by a slight excess (4%) of Ba. The sample was finally annealed at 1300 °C in oxygen (as for x = 0.02 we showed that oxygen annealing caused the VSR to disappear so that only FR stayed11). 4 represents practically the pure apatite phase and exhibits only FR behavior (see Fig. S10 and S11, ESI†). The dc susceptibility follows the Curie–Weiss law with θ = −3.2 K and μeff = 4.99 μB, which is close to 4.9 μB for S = 2, implying that the compound contains Co3+ with a high-spin d6 configuration (Fig. S12, ESI†). Rietveld refinement does not show an appreciable electron density near (0,0,0) where a Co atom would be found, while an abnormally low atomic displacement parameter for P suggests Co substitution on P sites (Table S7, ESI†).
Comparing the susceptibilities at 10 K we found that 10% of Co ions in 2 belong to the paramagnetic centers observed in 4. Using the χT(T) and low-temperature M(H) dependence measured for 2 and 4 (Fig. S13 and S14, ESI†) we corrected the data of 2 (Fig. 4). The corrected M(H) data at 1.8 K indicate the magnetization of VSR centers only.
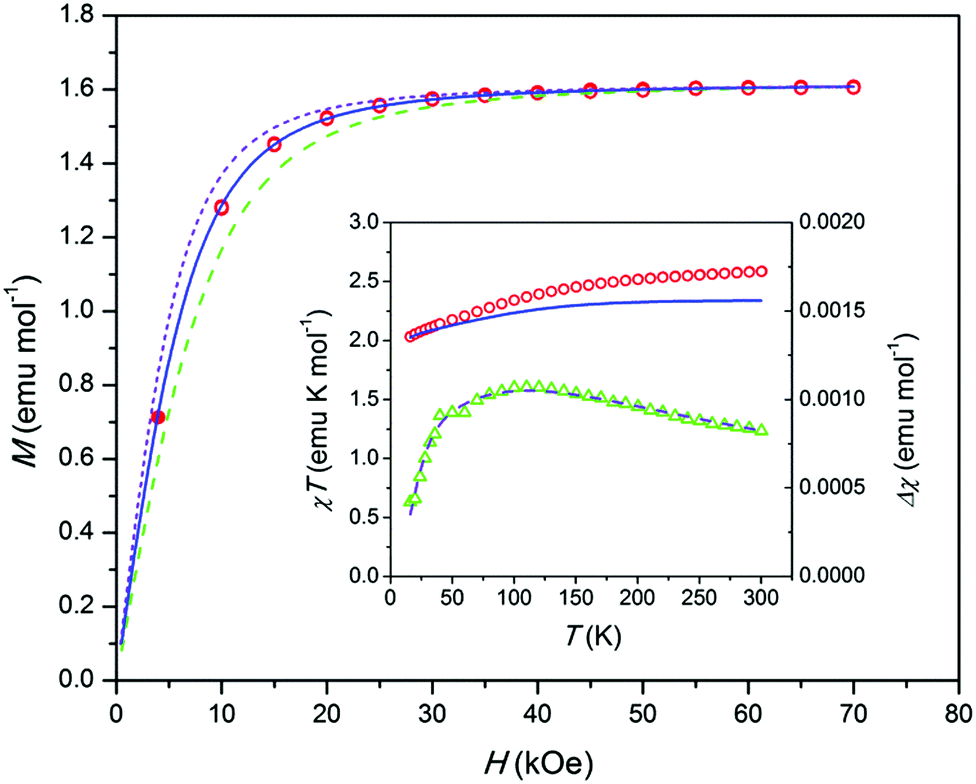 | ||
| Fig. 4 Sample 2. Magnetization M corrected for an FR contribution and normalized to 1 mol of Co vs. magnetic field H at T = 1.8 K. Open symbols – data from the reversible part of M(H), filled symbols – from an equilibrium dc value after the relaxation for 2 hours. Lines – modeling; dashed – gz = 8, solid – gz = 10, short dashed – gz = 12. Inset: Left axis, product χT corrected as above vs. temperature T. Circles – experimental data, solid line – modeling. Right axis, difference Δχ between the experimental and modeled χ vs. T. Triangles – experiment, dashed line – fitting as described in the text and ESI.† | ||
According to a modeling,11 an electronic structure of [OCoO] compatible with the observed high Ueff implies that the ground doublet has very high easy axis anisotropy with gx and gy close to zero. In this case the low-temperature M(H) may be approximately described by a Brillouin function with a certain gz and Seff = 1/2 and with a proper powder-averaging by a numerical integration.9 Then by fitting experimental data, one can get simultaneously both, gz = 2(L + 2S) and a content of paramagnetic centers. Our modeling shows that the corrected M(H) data fit well to gz = 10 with a content of paramagnetic centers corresponding to 58% of Co in 2 (see Fig. 4). Since such a quantity of Co can be found only in the apatite trigonal channels, this supports the assumption11 that the VSR center stems from an intrachannel Co atom like an NSR center. The remaining magnetically not-assessed 32% of Co ions seem to be in non-magnetic states at T = 1.8 K, e.g. as exchange-coupled ions or low-spin Co3+. To such ions, the CoO impurity obviously contributes.
The VSR center, an intra-channel twofold coordinated Co atom, most probably adopts an oxidation state of +2 or +3. Among appropriate ground electronic multiplets for the linear Co complex discussed in ref. 11, only 4Φg (Co2+) may provide large enough zero-field splitting to justify the observed Ueff values, also for a moderately bent [OCoO] group. By modeling using the program CONDON,16 with 4Φg as a starting point, and adjusting the ligand field parameters, we obtained a theoretical χT(T) dependence for a bent [OCoIIO] (Fig. 4 inset, solid line). It corresponds to an electronic state with a first excited doublet at an energy very close to an experimental Ueff of 338 cm−1 and with a gz = 10 ground doublet (see the ESI†). The calculated χT values approach the experimental ones at low temperature, but fall below as the temperature increases. Such a behavior may be connected to the presence of a fraction of antiferromagnetically coupled Co ions which do not contribute to the magnetization at low temperature but add an increasing quantity at increasing temperatures due to the decoupling of spins (this also concerns the CoO impurity). This assumption is further supported by the analysis of the difference Δχ(T) between experimental and calculated χ(T). The Δχ(T) data show a behavior common for an antiferromagnetic exchange cluster with the Δχ maximum at an intermediate temperature (Fig. 4 inset, triangles). It fits well to a Heisenberg S = 3/2 dimer and comprises ca. 13% of Co ions in 2. The fitting includes also a temperature independent paramagnetic contribution χTIP. It may account for the CoO admixture, the value being underestimated though (ca. 4% of Co ions in 2). The proposed dimers imply that some Co atoms may form intra-channel groups [OCoOCoO], which is allowed by the size of the channel.
As a result for 2, the following picture emerges. A major fraction of Co ions situated in the trigonal channels form the VSR centers and a minor fraction of the Co ions form the exchange-coupled clusters (probably dimers). Together they comprise 71% of Co ions, the number approaching that (74%) derived from the intra-channel crystallographic occupancy. The VSR center includes a moderately bent atomic group [OCoO] apparently different from an earlier identified NSR center, a dioxocobaltate(II) ion. However, the electronic structure modeling suggests that the VSR center contains Co2+. Hence one may suppose that only next atoms in the vicinity of the Co atom are different for these two types of paramagnets. For example, the VSR center may represent hypothetical oxoperoxocobaltate(II) or oxohydroxocobaltate(II) ions. Of course, this proposal needs further clarification. The FR centers may be attributed mostly to crystallographically non-assessed Co ions in oxidation state +3.
In summary, by Co doping, two types of SIM with high concentration can be controllably realized in an apatite lattice: the first one with a moderate Ueff11 and the second one with an exceptionally high Ueff. As mentioned previously in the context of materials showing moderate Ueff,11 upon further increase of the Co content, an ordered hexagonal array of SIMs might be formed on the apatite (001) crystal surface with the grid interspacing equal to the cell parameter a. Having in mind that the high-Ueff SIMs are also located in the apatite channels and in a high concentration, the same attractive prospects apply for the latter. Noteworthily, Ueff increases by a factor of 7 on going from the first to the second type SIM. This strongly enhances chances for this type of material to be a prototype for an atomic-scale magnetic memory.
The work was supported by the Russian Science Foundation (RSF) under Grant no. 16-13-10031. We thank Walter Schnelle and Eva Bruecher for help with magnetic measurements and Christine Stefani for the XRD measurements.
Notes and references
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- L. Ungur and L. F. Chibotaru, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., 2016, 128, 16305–16308 CrossRef.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, J. R. Long, M. Atanasov, F. Neese, F. Grandjean and G. J. Long, Inorg. Chem., 2013, 52, 13123–13131 CrossRef CAS PubMed.
- A. A. Pavlov, Y. V. Nelyubina, S. V. Kats, L. V. Penkova, N. N. Efimov, A. O. Dmitrienko, A. V. Vologzhanina, A. S. Belov, Y. Z. Voloshin and V. V. Novikov, J. Phys. Chem. Lett., 2016, 7, 4111–4116 CrossRef CAS PubMed.
- A. Jesche, R. W. McCallum, S. Thimmaiah, J. L. Jacobs, V. Taufour, A. Kreyssig, R. S. Houk, S. L. Bud'ko and P. C. Canfield, Nat. Commun., 2014, 5, 3333 CAS.
- I. G. Rau, S. Baumann, S. Rusponi, F. Donati, S. Stepanow, L. Gragnaniello, J. Dreiser, C. Piamonteze, F. Nolting, S. Gangopadhyay, O. R. Albertini, R. M. Macfarlane, C. P. Lutz, B. A. Jones, P. Gambardella, A. J. Heinrich and H. Brune, Science, 2014, 344, 988–992 CrossRef CAS PubMed.
- P. E. Kazin, M. A. Zykin, Y. V. Zubavichus, O. V. Magdysyuk, R. E. Dinnebier and M. Jansen, Chem. – Eur. J., 2014, 20, 165–178 CrossRef CAS.
- P. E. Kazin, M. A. Zykin, W. Schnelle, C. Felser and M. Jansen, Chem. Commun., 2014, 50, 9325–9328 RSC.
- P. E. Kazin, M. A. Zykin, W. Schnelle, Y. V. Zubavichus, K. A. Babeshkin, V. A. Tafeenko, C. Felser and M. Jansen, Inorg. Chem., 2017, 56, 1232–1240 CrossRef CAS PubMed.
- P. E. Kazin, O. R. Gazizova, A. S. Karpov, M. Jansen and Y. D. Tretyakov, Solid State Sci., 2007, 9, 82–87 CrossRef CAS.
- S. M. J. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L.-C. Brunel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 1999, 38, 5329–5340 CrossRef CAS.
- M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC.
- P. S. Silinsky and M. S. Seehra, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 419–423 CrossRef CAS.
- H. Schilder and H. Lueken, J. Magn. Magn. Mater., 2004, 281, 17–26 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, powder X-ray diffraction patterns, magnetic and relaxation data plots, crystal structure data, modeling and fitting data. See DOI: 10.1039/c7cc02453c |
| ‡ Further details of the crystal structure investigations may be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (Fax: +49-7247-808-666; E-mail: crysdata@fizkarlsruhe.de, http://www.fiz-karlsruhe.de/request for deposited data.html) on quoting the depository number CSD 432190. |
| This journal is © The Royal Society of Chemistry 2017 |