
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkynyl sulfoxides as α-sulfinyl carbene equivalents: gold-catalysed oxidative cyclopropanation†
Matthew J.
Barrett
,
Ghulam F.
Khan
,
Paul W.
Davies
 * and
Richard S.
Grainger
*
* and
Richard S.
Grainger
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: p.w.davies@bham.ac.uk; r.s.grainger@bham.ac.uk
First published on 4th May 2017
Abstract
Alkynyl sulfoxides are shown to act as α-sulfinyl metallocarbene synthons under oxidative gold catalysis, enabling reactions that are not available from diazo-precursors. This strategy is exemplified in the synthesis of fused α-sulfinyl cyclopropanes.
Metallocarbenes underpin a broad range of powerful chemo- and stereoselective transformations in modern organic synthesis.1 α-Sulfinyl metal carbenes, 1, position a readily-elaborated functional group2–5 bearing a stereogenic centre at the reactive site (Scheme 1a).6 However this attractive proposition has yet to be realised using conventional approaches to metal carbene reactivity. Maguire and co-workers established that α-diazo sulfoxides are only isolable when constrained as part of a cyclic system and that they and their resulting α-sulfinyl rhodium carbenes undergo rapid Wolff-like rearrangement (Scheme 1b).7 Here we demonstrate how the reactivity patterns of α-sulfinyl carbenes can be accessed from alkynyl sulfoxides under gold catalysis.
 | ||
| Scheme 1 Approaches to access α-sulfinyl metal carbene type reactivity. | ||
The use of a π-acid to chemoselectively activate alkynes in the presence of a nucleophilic oxidant provides an attractive route into α-oxo metal carbene reactivity patterns without the need to install or handle diazo groups.8–11 One intriguing aspect is that some reactions appear to bypass the actual gold carbene 4 and proceed directly from the vinyl gold carbenoid intermediate 3 (Scheme 1c).12 We hypothesised that broader applications of α-sulfinyl metal carbene chemistry might therefore be accessible if α-sulfinyl vinyl gold carbenoid 7 could be accessed from alkynyl sulfoxide 6, quenched prior to expulsion of the nucleofuge, and proved less vulnerable to rearrangement than the corresponding metal carbene. This approach presents an interesting challenge as sulfoxides are effective nucleophiles and oxygen-transfer agents in the presence of alkyne–gold complexes13 or metal carbenes.14 For successful application of alkynyl sulfoxide 6 as an α-sulfinyl carbene equivalent, effective π-activation and regioselective oxidation is required, but 6 and 8 must not act as nucleophilic oxidants.15
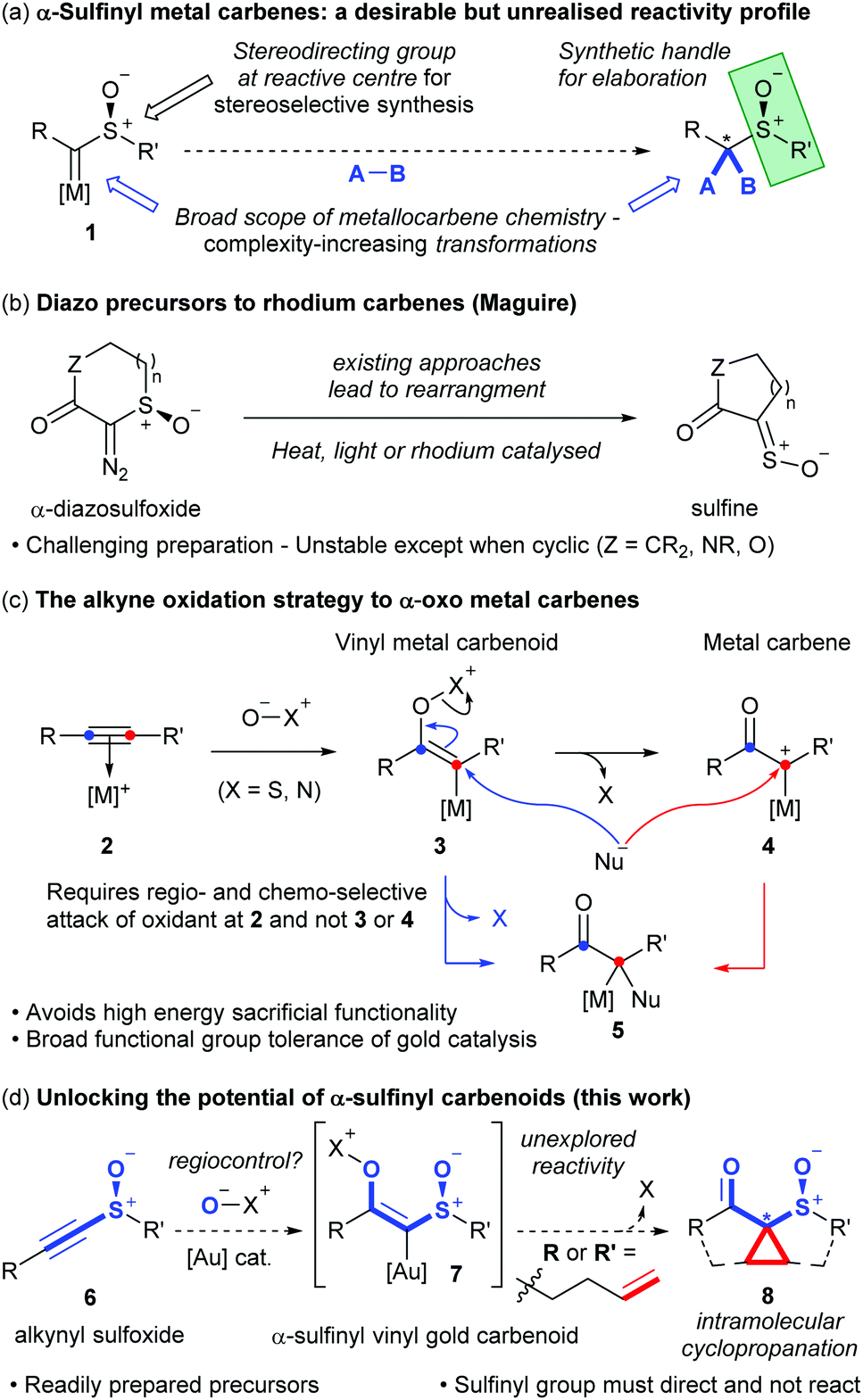
We tested this hypothesis in the oxidative cyclopropanation reaction of readily accessible ene-alkynyl sulfoxides.‡ A reaction survey with 9a identified that the desired cyclopropane-fused thiolane S-oxide was formed as an approximately 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers 10a and 10b using 3,5-dichloropyridine-N-oxide (11) as stoichiometric oxidant in the presence of various cationic Au(I) catalysts. Phosphite, N-heterocyclic carbene and bulky phosphine ligands all proved effective on the gold, with SPhosAuNTf2 giving highest yield (Table 1, entries 1–5). Dioxane proved superior to other solvents (entries 5–9) while 11 was more effective than other commonly used pyridine-N-oxide derivatives 12 and 13 (entries 10–13).16 Changing the temperature had little effect on dr, though conversion stalled at much lower temperatures: at 80 °C the catalyst loading could be halved with little effect, though dropping further was detrimental to conversion of 9a (entry 10). Increasing oxidant loading saw lower yields, likely due to over-oxidation pathways (entry 11).
:1 mixture of diastereomers 10a and 10b using 3,5-dichloropyridine-N-oxide (11) as stoichiometric oxidant in the presence of various cationic Au(I) catalysts. Phosphite, N-heterocyclic carbene and bulky phosphine ligands all proved effective on the gold, with SPhosAuNTf2 giving highest yield (Table 1, entries 1–5). Dioxane proved superior to other solvents (entries 5–9) while 11 was more effective than other commonly used pyridine-N-oxide derivatives 12 and 13 (entries 10–13).16 Changing the temperature had little effect on dr, though conversion stalled at much lower temperatures: at 80 °C the catalyst loading could be halved with little effect, though dropping further was detrimental to conversion of 9a (entry 10). Increasing oxidant loading saw lower yields, likely due to over-oxidation pathways (entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T (°C) | Oxidant | Yield of 10aa (%) |
a Reactions performed on a 0.1 mmol scale; yields of the major diastereomer 10a determined by 1H NMR analysis of the crude reaction mixture using 1,2,4,5-tetramethylbenzene as an internal reference. Overlap prevented accurate determination of dr.
b 62% at 2.5 mol% cat. 27% at 1.0 mol% cat. L1 = (tris(2,4-di-tertbutylphenyl)phosphite). L2 = 1,3-bis(2,6-diisopropylphenyl-imidazol-2-ylidene). L3 = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl. L4 = 2-ditertbutylphosphinobiphenyl. L5 = 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos).
c Higher concentrations afforded lower yields (42% 10a at 0.2 M).
|
|||||
| 1 | L1 | Dioxane | 65 | 11 | 42 |
| 2 | L2 | Dioxane | 65 | 11 | 45 |
| 3 | L3 | Dioxane | 65 | 11 | 47 |
| 4 | L4 | Dioxane | 65 | 11 | 59 |
| 5 | L5 | Dioxane | 65 | 11 | 66 |
| 6 | L5 | 1,2-DCE | 60 | 11 | 45 |
| 7 | L5 | THF | 60 | 11 | 37 |
| 8 | L5 | CH2Cl2 | rt | 11 | 26 |
| 9 | L5 | Toluene | 60 | 11 | 39 |
| 10 | L5 | Dioxane | 80 | 11 | 69b,c |
| 11 | L5 | Dioxane | 80 | 11 (2.0 eq.) | 54 |
| 12 | L5 | Dioxane | 80 | 12 | 63 |
| 13 | L5 | Dioxane | 80 | 13 | 59 |
A range of ene-alkynyl sulfoxides 9a–v were prepared to explore the effect of the alkyne substituent on the reaction (Table 2). Primary, secondary and tertiary alkyl substituents were all accommodated with good conversions at 50 °C (entries 1–6). Notably, cyclopropyl-substituted alkyne 3q gave the same yield and d.r. at room temperature (entry 6). Aryl substituted alkynes were also more reactive, proceeding at room temperature, although higher yields were obtained under the standard conditions (entries 7–20, see ESI† for reactions at room temperature). In these cases the d.r. was approximately 8:1 as determined by 1H NMR analysis of the reaction mixture before purification. The aromatic substituent can be either electron-rich or -poor and will accommodate a variety of functionality across all positions. The tolerance of this chemistry is highlighted by the ready inclusion of a 3-bromothiophen-2-yl moiety (entry 20). Furthermore, the reactions of diene-alkynyl sulfoxides 9u/v proceeded smoothly to the desired sulfur heterocycles despite the possibility of competing cycloisomerisation prior to oxidation across one or both of the two 1,6-enyne motifs embedded in the substrates (entries 21 and 22).17
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | 9 | T (°C) | Time (h) | % Yield of 10a |
|
a Isolated yields after purification by column chromatography. The yields refer to a single diastereomer apart from when diastereomeric ratios are given.
b
9c is a 1:1 mixture of diastereomers.
c Incomplete conversion.
d The same yield and d.r. were obtained at room temperature.
e 2.5 mol% SPhosAuNTf2.
|
|||||
| 1 | n Bu | 9a | 50 | 17 | 72 (6:1) |
| 2 | PhCH2CH2 | 9b | 50 | 3.5 | 63 (6:1) |
| 3 |

|
9c | 50 | 21 | 70 (10:10:1:1) |
| 4 | Cyclohexyl | 9d | 50 | 24c | 45 (7:1) |
| 5 | t Bu | 9e | 50 | 25 | 70 (12:1) |
| 6 | Cyclopropyl | 9f | 50 | 17 | 86d (10:1) |
| 7 | Ph | 9g | 65 | 0.75 | 80 |
| 8e | 4-MeC6H4 | 9h | 23 | 28 | 75 |
| 9 | 4-MeOC6H4 | 9i | 40 | 1 | 78 |
| 10e | 4-AcNH-C6H4 | 9j | 50 | 20 | 79 |
| 11 | 4-F3CC6H4 | 9k | 50 | 17 | 63 |
| 12 | 4-MeO2CC6H4 | 9l | 50 | 20 | 64 |
| 13 | 4-FC6H4 | 9m | 50 | 17 | 68 |
| 14 | 3-MeOC6H4 | 9n | 50 | 18 | 70 |
| 15 | 4-BrC6H4 | 9o | 50 | 3 | 74 |
| 16e | 2-BrC6H4 | 9p | 23 | 28 | 50 |
| 17 | 2-iPr-C6H4 | 9q | 50 | 28 | 52 (7:1) |
| 18 | 2-Naphthyl | 9r | 50 | 28 | 70 |
| 19 | 2-Furyl | 9s | 50 | 28 | 74 |
| 20 |

|
9t | 50 | 28 | 73 |
| 21 |
|
9u | 50 | 21 | 63 (10:1) |
| 22 |

|
9v | 50 | 21 | 65 (10:1) |

The relative stereochemistry of the major diastereomers 10 and minor diastereomers 10′ were assigned using characteristic chemical shifts in the 1H NMR spectra (see ESI†).
In addition a crystal structure was obtained for major diastereomer 10g (Fig. 1),§ confirming the NMR analysis that the sulfoxide oxygen and cyclopropyl methylene are on the same side of the thiolane ring.
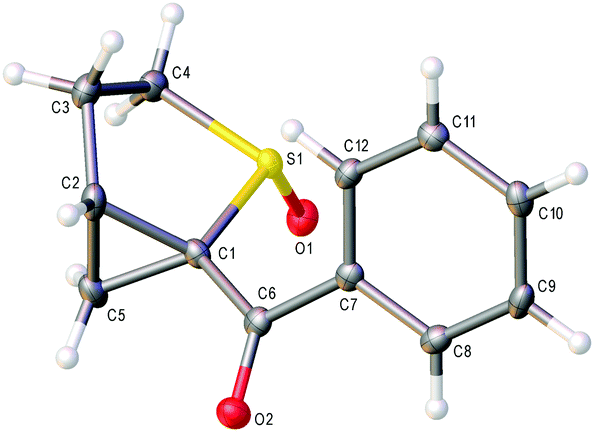 | ||
| Fig. 1 X-ray crystal structure of major diastereomer 10g. | ||
The reaction of 9q, bearing an ortho-isopropyl substituent, saw formation of a side-product alongside 10q (Table 2, entry 17) although this was not isolated in sufficient quantity or purity to allow full characterisation. We hypothesised that 1,5-hydride transfer from the benzylic position may be competing with cyclopropanation.18 To test this hypothesis we prepared the methylsulfoxide 12 where cyclopropanation is not possible. The formation of stilbene 13 under the standard reaction conditions is indeed consistent with 1,5-hydride transfer onto a vinyl gold carbenoid (cf.7) followed by elimination of a proton and protodeauration (Scheme 2). Key resonances in 13 also correlate to those in the side-product from 9q.
 | ||
| Scheme 2 An alternative reaction pathway consistent with 1,5-hydride transfer. | ||
The feasibility of using a disubstituted alkene in the cyclopropanation was then explored using styrene 14 (Scheme 3). Under the standard reaction conditions the more heavily substituted cyclopropane 15 was indeed formed,19 alongside hydroxylated ring-opened product 16. Formation of 16 is consistent with the cationic character of a gold carbenoid extending through the alkene and enabling a hydrative cyclisation in the presence of adventitious water.20
 | ||
| Scheme 3 Use of 1,2-disubstituted alkene in oxidative cyclopropanation. | ||
A preliminary investigation shows that using alkynyl sulfoxides as α-sulfinyl carbene equivalents is not limited to sulfur heterocycle formation. Under unoptimised conditions, which saw incomplete conversion, 1,5-enyne 17 gave fused carbocyclic ring system 18 as a 1.6:1 mixture of diastereomers (Scheme 4).
 | ||
| Scheme 4 Synthesis of an α-sulfinyl cyclopropyl-fused cyclopentanone. | ||
In conclusion, the synthetic limitations that have prevented access to desirable aspects of α-sulfinyl metallocarbene reactivity can be bypassed by an oxidative gold catalysis strategy using readily accessed alkynyl sulfoxides. For the first time α-sulfinyl carbene-like activity is demonstrated through intramolecular cyclopropanation reactions, affording ring-fused cyclopropanes containing α-sulfinylcarbonyl motifs.21 Future work will address the use of this approach in the wider context of carbene reactivity and explore the opportunities arising from the use of enantiopure sulfoxides.22
The authors acknowledge support from the Centre for Chemical and Materials Analysis in the School of Chemistry at University of Birmingham (UoB) and thank Dr Louise Male (UoB) for X-ray crystallography. We thank the UoB for a studentship (MJB).
Notes and references
- Carbene Chemistry, ed. G. Bertrand, Fontis Media, Lausanne, Switzerland, 2002 Search PubMed.
- For a review of the Pummerer reaction: L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832–5844 CrossRef CAS PubMed.
- For reviews of elimination reactions of sulfoxides, see: (a) A. L. Schwan, ChemCatChem, 2015, 7, 226–227 CrossRef CAS; (b) M. C. Aversa, A. Barattucci, P. Bonaccorsi and P. Giannetto, Curr. Org. Chem., 2007, 11, 1034–1052 CrossRef CAS . For prior work from one of us, see; (c) S. T. Bedford, R. S. Grainger, J. W. Steed and P. Tisselli, Org. Biomol. Chem., 2005, 3, 404–406 RSC; (d) R. S. Grainger, P. Tisselli and J. W. Steed, Org. Biomol. Chem., 2004, 2, 151–153 RSC.
- For recent representative examples of metal-sulfoxide exchange, see: (a) W. M. Dean, M. Šiaučiulis, T. E. Storr, W. Lewis and R. A. Stockman, Angew. Chem., Int. Ed., 2016, 55, 10013–10016 CrossRef CAS PubMed; (b) P. J. Rayner, P. O’Brien and R. A. J. Horan, J. Am. Chem. Soc., 2013, 135, 8071–8077 CrossRef CAS PubMed; (c) M. Hughes, T. Boultwood, G. Zeppetelli and J. A. Bull, J. Org. Chem., 2013, 78, 844–854 CrossRef CAS PubMed.
- For a recent review of sulfoxide-directed C–H functionalization, see: A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS PubMed.
- For reviews on the application of enantiopure sulfoxides, see: (a) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed; (b) M. C. Carreño, G. Hernández-Torres, M. Ribagorda and A. Urbano, Chem. Commun., 2009, 6129–6144 RSC.
- (a) P. G. McCaw, N. M. Buckley, K. S. Eccles, S. E. Lawrence, A. R. Maguire and S. G. Collins, J. Org. Chem., 2017, 82, 3666–3679 CrossRef CAS PubMed; (b) O. C. M. O’Sullivan, S. G. Collins, A. R. Maguire and G. Bucher, Eur. J. Org. Chem., 2014, 2297–2304 CrossRef; (c) S. G. Collins, O. C. M. O’Sullivan, P. G. Kelleher and A. R. Maguire, Org. Biomol. Chem., 2013, 11, 1706–1725 RSC; (d) O. C. M. O’Sullivan, S. G. Collins and A. R. Maguire, Synlett, 2008, 659–662 Search PubMed; (e) O. C. M. O’Sullivan, S. G. Collins, A. R. Maguire, M. Böhm and W. Sander, Eur. J. Org. Chem., 2006, 2918–2924 CrossRef; (f) A. R. Maguire, S. G. Collins and A. Ford, ARKIVOC, 2003, 7, 96–109 Search PubMed; (g) W. Sander, A. Strehl, A. R. Maguire, S. Collins and P. G. Kelleher, Eur. J. Org. Chem., 2000, 3329–3335 CrossRef CAS; (h) A. R. Maguire, P. G. Kelleher and S. E. Lawrence, Tetrahedron Lett., 1998, 39, 3849–3852 CrossRef CAS; (i) A. R. Maguire, P. G. Kelleher, G. Ferguson and J. F. Gallagher, Tetrahedron Lett., 1998, 39, 2819–2822 CrossRef CAS.
- For reviews, see: (a) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016, 45, 4448–4458 RSC; (b) H.-S. Yeom and S. Shin, Acc. Chem. Res., 2014, 47, 966–977 CrossRef CAS PubMed.
- For examples in oxidative cyclopropanation, see: (a) H. Chen and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11775–11779 CrossRef CAS PubMed; (b) K. Ji, Z. Zheng, Z. Wang and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1245–1249 CrossRef CAS PubMed; (c) H.-S. Yeom and S. Shin, Org. Biomol. Chem., 2013, 11, 1089–1092 RSC; (d) A. Homs, M. E. Muratore and A. M. Echavarren, Org. Lett., 2015, 17, 461–463 CrossRef CAS PubMed; (e) D. Qian, H. Hu, F. Liu, B. Tang, W. Ye, Y. Wang and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 13751–13755 CrossRef CAS PubMed; (f) K.-B. Wang, R.-Q. Ran, S.-D. Xiu and C.-Y. Li, Org. Lett., 2013, 15, 2374–2377 CrossRef CAS PubMed; (g) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 50, 6911–6914 CrossRef CAS PubMed; (h) D. Qian and J. Zhang, Chem. Commun., 2011, 47, 11152–11154 RSC and references therein.
- For prior work in this area from one of us, see: (a) M. Dos Santos and P. W. Davies, Chem. Commun., 2014, 50, 6001–6004 RSC; (b) P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379–381 RSC; (c) P. W. Davies, Pure Appl. Chem., 2010, 82, 1537–1544 CrossRef CAS; (d) P. W. Davies and S. J. C. Albrecht, Angew. Chem., Int. Ed., 2009, 48, 8372–8375 CrossRef CAS PubMed.
- Review of alkyne π-acid activations: A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed.
- For examples, see: (a) Ref. 10a ; (b) S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 2939–2942 CrossRef CAS PubMed.
- Recent examples (a) T. Matsuda, Y. Nishida, K. Yamanaka and Y. Sakurai, Tetrahedron, 2015, 71, 869–874 CrossRef CAS; (b) M. J. Barrett, P. W. Davies and R. S. Grainger, Org. Biomol. Chem., 2015, 13, 8676–8686 RSC; (c) H.-S. Yeom and S. Shin, Org. Biomol. Chem., 2013, 11, 1089–1092 RSC; (d) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512–8524 CrossRef CAS PubMed and references therein.
- See ref. 13d and C. A. Witham, P. Mauleon, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS PubMed.
- Alkynyl sulfoxides have been scarcely employed in gold catalysis. For a single example, see G. Zhang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 12598–12599 CrossRef CAS PubMed.
- (a) D. B. Huple, S. Ghorpade and R.-S. Liu, Chem. – Eur. J., 2013, 19, 12965–12969 CrossRef CAS PubMed; (b) R. Liu, G. N. Winston-McPherson, Z.-Y. Yang, X. Zhou, W. Song, I. A. Guzei, X. Xu and W. Tang, J. Am. Chem. Soc., 2013, 135, 8201–8204 CrossRef CAS PubMed.
- For a review, see: R. Dorel and A. M. Echavarren, J. Org. Chem., 2015, 80, 7321–7332 CrossRef CAS PubMed.
- In keeping with the oxidative cyclisation of o-benzylalkynes, where C–C bond formation precedes the elimination observed here, see ref. 12b.
- The methylene protons on the non-fused cyclopropane in 15 are desymmetrised to 4 distinct resonances, consistent with an interaction with the superimposed phenyl group in an exo position and retention of starting alkene geometry.
- For example of alkoxycyclisation of enynes in the presence of water C. Nieto-Oberhuber, M. P. Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem. – Eur. J., 2006, 12, 1677–1693 CrossRef CAS PubMed.
- All the reaction outcomes are consistent with the sequence of sulfinyl-directed oxidation followed by cyclopropanation. For instance, an inverted order of steps would be expected to give rise to vinylcyclopropanes through fast 1,2-CH insertion from alkylalkynes.
- Although not the focus of this report, analogous cyclopropane-fused sulfolanes can be prepared under the reported reaction conditions starting from the alkynyl sulfone equivalent.
For examples of gold-catalysed oxidative transformations of alkynyl sulfones see: L. Cui, G. Zhang, Y. Peng and L. Zhang, Org. Lett., 2009, 11, 1225–1228 CrossRef CAS PubMed and ref. 9b.

Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data for new compounds. 1H and 13C NMR spectra. Structural determination and additional catalysis results. CCDC 1528851. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc02244a |
| ‡ All sulfoxides were prepared in the racemic series. |
§ Crystal structure determination of 10g: crystal data for C12H12O2S (M = 220.28 g mol−1): triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 6.2782(3) Å, b = 7.1917(3) Å, c = 12.4920(6) Å, α = 86.275(4)°, β = 75.966(4)°, γ = 66.086(4)°, V = 499.87(4) Å3, Z = 2, T = 100.01(11) K, μ(CuKα) = 2.667 mm−1, Dcalc = 1.463 g cm−3, 7653 reflections measured (7.3° ≤ 2Θ ≤ 144.236°), 1939 unique (Rint = 0.0218, Rsigma = 0.0170) which were used in all calculations. The final R1 was 0.0390 (I > 2σ(I)) and wR2 was 0.0963 (all data). The CIF for the crystal structure of 10g has been deposited with the CCDC and have been given the deposition number CCDC 1528851. (no. 2), a = 6.2782(3) Å, b = 7.1917(3) Å, c = 12.4920(6) Å, α = 86.275(4)°, β = 75.966(4)°, γ = 66.086(4)°, V = 499.87(4) Å3, Z = 2, T = 100.01(11) K, μ(CuKα) = 2.667 mm−1, Dcalc = 1.463 g cm−3, 7653 reflections measured (7.3° ≤ 2Θ ≤ 144.236°), 1939 unique (Rint = 0.0218, Rsigma = 0.0170) which were used in all calculations. The final R1 was 0.0390 (I > 2σ(I)) and wR2 was 0.0963 (all data). The CIF for the crystal structure of 10g has been deposited with the CCDC and have been given the deposition number CCDC 1528851. |
| This journal is © The Royal Society of Chemistry 2017 |