
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective CO2vs. N2 adsorption in the cavity of a molecular coordination cage†
James S.
Wright‡
,
Alexander J.
Metherell‡
,
William M.
Cullen
 ,
Jerico R.
Piper
,
Robert
Dawson
* and
Michael D.
Ward
*
,
Jerico R.
Piper
,
Robert
Dawson
* and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: m.d.ward@sheffield.ac.uk; r.dawson@sheffield.ac.uk
First published on 31st March 2017
Abstract
Two M8L12 cubic coordination cages, as desolvated crystalline powders, preferentially adsorb CO2 over N2 with ideal selectivity CO2/N2 constants of 49 and 30 at 298 K. A binding site for CO2 is suggested by crystallographic location of CS2 within the cage cavity at an electropositive hydrogen-bond donor site, potentially explaining the high CO2/N2 selectivity compared to other materials with this level of porosity.
Porous solid-state materials are attractive for gas adsorption purposes, with several classes of porous material gaining increasing attention in recent years. These include metal–organic frameworks (MOFs)/coordination polymers;1–17 covalent organic frameworks (COFs)/microporous organic polymers (MOPs);18–24 molecular cages;25–35 and other molecular crystals.36–43
In the case of MOFs and MOPs, impressive gas uptake capacities have been reported, and extremely highly porous materials described.12,14,21 However, higher uptake capacity in porous materials can come at the expense of selectivity between small gaseous molecular guests, as shown in previous work comparing porous organic cages of different pore sizes with each other, and with MOFs.32 Adsorbents which are selective for the desired adsorbate are desirable, but not necessarily at the expense of uptake capacity. For this purpose, the design of flexible adsorbents whose pores may open under the influence of an external stimulus has been demonstrated, both in MOFs1,7,13 and extrinsically porous materials;17,37,42 this is still an emerging field.
Perhaps better developed is the functionalisation of the pore space of intrinsically porous materials, to enhance selectivity for binding of different gaseous guests. In particular, the improvement of CO2 adsorption selectivity in MOFs has been demonstrated by the addition of hydrogen-bonding sites3,11 or the fluorination of pores.5,44,45 These internal surface modifications can however come at the expense of uptake capacity by occupying some of the interior space, so an adsorbent in which a binding site is built into the ‘walls’ of the cavity is desirable.
We have previous reported the structures and guest binding properties of the cubic coordination cages [M8L12]X16, in which M are transition metal dications [usually Co(II)] located at the vertices of the cage, and L are bis(pyrazolyl-pyridine) bridging ligands which connect a pair of metal ions along every edge of the assembly (Fig. 1).46–52 The ligand L may be unsubstituted (Lo: R = H in the figure) in which case the cages are soluble in polar organic solvents;47,48 or may be substituted (Lw: R = CH2OH in the figure) to make the cages water-soluble.49–52 These cages have been shown to bind a wide range of organic guests in the central cavity. In organic solvents guest binding is partly driven by hydrogen-bonding of electron-rich regions of guests to H-bond donor pockets located on the interior surface of the cage, in regions of high positive electrostatic potential; this affords binding constants in the range 102–103 M−1.47 In water, the hydrophobic effect provides the dominant driving force for strong binding of hydrophobic guests with binding constants of up to 108 M−1.49–51 Here we report an investigation into the gas sorption capability of these materials, demonstrating a high selectivity for CO2 uptake over N2 in the solid state, which we ascribe to the presence of the same H-bond donor sites on the cage interior surface that facilitate guest binding in solution.47,53
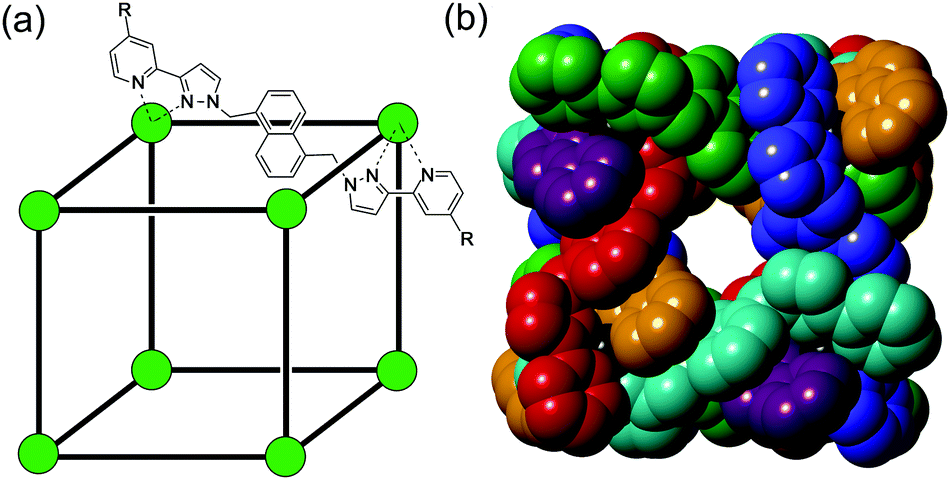 | ||
| Fig. 1 General structure of host cages [M8L12](BF4)16: types A (R = H, M = Cd) and B (R = CH2OH, M = Co). (a) A sketch emphasising the cubic array of M(II) ions and the disposition of a bridging ligand spanning an edge; (b) a space-filling representation of the complex cation with ligands coloured separately for clarity (see ref. 46–53). | ||
We used the cages [Cd8(Lo)12](BF4)16 (A)48 and [Co8(Lw)12] (BF4)16 (B),49 both of which have been reported before. The compounds were prepared as methanol solvates, and then dried and thermally desolvated. Powder X-ray diffraction analysis of the dried materials (ESI†) showed that B retains the same phase that was observed in the single crystal structure of the methanol solvate, whereas A loses crystallinity and becomes amorphous. This is likely related to the fact that in B the presence of hydroxymethyl groups on the exterior surface of the cages results in a formation of an intermolecular O–H⋯O hydrogen-bonding network of cage molecules which allows crystallinity to be retained even when solvent molecules are lost (ESI†). In A in contrast there are no such interactions between the exterior surfaces of adjacent cages and crystallinity is lost on desolvation. However, 1H NMR and mass spectrometric analyses confirmed that the integrity of the molecular cages is retained even when the crystals are desolvated.
The cages were found to be non-porous having BET surface areas <20 m2 g−1. The volumetric gas sorption isotherms were measured for uptake of CO2 and N2 by both cages at 298 K (Fig. 2) and also at 273 K (ESI†). Both cages demonstrate highly selective uptake for CO2vs. N2 at both temperatures. The gas uptake comparisons and selectivity constants are summarised in Table 1, and Henry's law calculation data is presented in the ESI.† The capacity for CO2 uptake is very similar for both cages. Although the cages have identical internal cavities, as we mentioned above the supramolecular structure of the cages is different because of the presence (cage B) or absence (cage A) of inter-cage hydrogen-bonding interactions between peripheral functional groups. This suggests either that CO2 uptake in the interstitial spaces between cages is very low, or that the void space between cages is similar in both materials (which in the case of the desolvated cage B is known to be small due to the hydrogen bonding, and therefore uptake here would be low anyway). We have noted in previous work that when crystalline cage samples are soaked in solutions of guests, quite large guest molecules can permeate the crystals and enter the cage cavities,50,52 even when the windows are occluded in the crystal structure and when the guest dimensions are larger than the 4 Å windows47 in the cage faces. Thus, for guest molecules as small as N2 or CO2, differences in crystal packing are unlikely to prevent adsorption: the similarity in CO2 uptake for both cage types therefore most plausibly relates to the similarity of the cavity inside cages A and B.49 Adsorption/desorption of CO2 is reversible in both cases with only a slight hysteresis, as is common for porous materials, which diminishes at low pressures.
 | ||
| Fig. 2 Volumetric gas sorption profiles for CO2 and N2 in cages A (top) and B (bottom) at 298 K. Filled circles represent adsorption and hollow circles represent desorption. | ||
| Cage | T (K) | 1 bar CO2 uptake (mmol g−1) | 1 bar N2 uptake (mmol g−1) | CO2/N2 selectivity constant | |
|---|---|---|---|---|---|
| Simple (ideal) | Henry's law | ||||
| B | 273 | 1.003 | 0.0309 | 32 | 156 |
| 298 | 0.672 | 0.0138 | 49 | 165 | |
| A | 273 | 1.005 | 0.027 | 37 | 59 |
| 298 | 0.673 | 0.022 | 30 | 32 | |
Attempts to locate CO2 guests within the cages were made using X-ray crystallography on single crystals under a CO2 atmosphere, but the crystals fractured rapidly into microcrystalline powder upon desolvation. Instead, using the method that has worked with other guests, single crystals of B (still solvated to prevent cracking) were soaked in liquid CS2 – as a structural analogue of CO2 – at 40 °C for 2 hours. This resulted in uptake of CS2 into the cage cavity.
Crystallographic analysis (ESI†) showed the structure to be [Co8(Lw)12](BF4)16·CS2·5H2O (Fig. 3) in which a molecule of CS2 is located such that it interacts with one of the hydrogen-bond donor pockets on the interior surface which are located at the two fac tris-chelate sites at either end of the long diagonal of the approximately cubic assembly.47–49
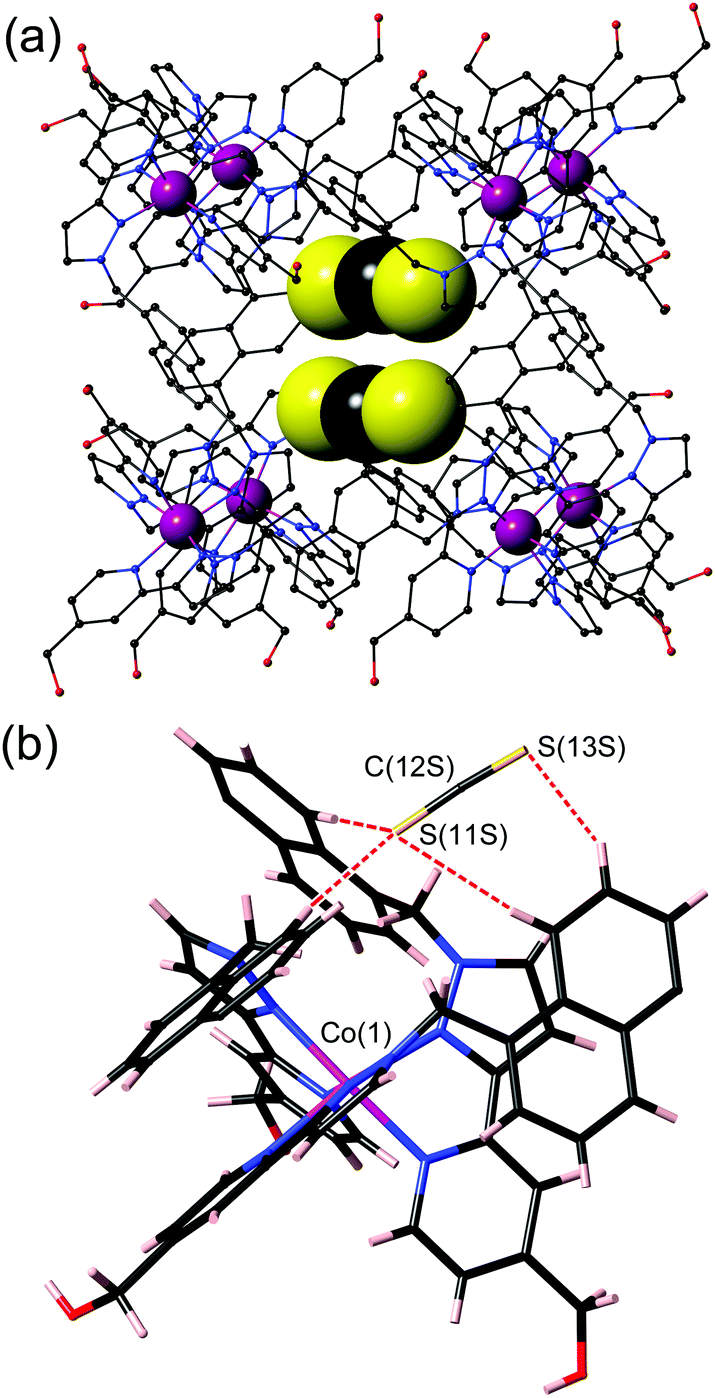 | ||
| Fig. 3 Crystal structure of the complex cation of [Co8(Lw)12](BF4)16·CS2·5H2O: (a) a view of the entire cage (in wireframe) with the CS2 guest (50% site occupancy in each of two positions) shown space-filling; (b) a close-up of the hydrogen-bonding environment around the CS2 guest, with the shortest CH⋯S contacts shown with red dashed lines. | ||
The site occupancy of the CS2 in each of the two pockets is 0.5, i.e. in the crystal structure there is one guest molecule per cage but it is disordered equally over the two possible sites. The CS2 guest is oriented such that the S atom [S(11S)] that is directed into the corner pocket is involved in several CH⋯S contacts (C⋯S distances in the range 3.5–3.7 Å) with H atoms from CH2 groups and naphthyl groups that converge around the guest binding site; the (non-bonded) Co(1)⋯S(11S) separation is 5.65 Å. The other S atom of the guest S(13S) is also involved in a short CH⋯S contact (3.51 Å) with an inwardly-directed naphthyl CH proton. This set of interactions is emphasised in Fig. 3(b).
The quadrupole moment of CS2 is opposite in sign to that of CO2 so in terms of point charges it is denoted (δ+)–(δ−)–(δ+);55,56 in this respect CS2 is not electronically analogous to CO2 although it is a reasonable geometric model. Nonetheless the ability of the S atoms of CS2 to act as hydrogen-bond acceptors, based on the local electron density at the S atoms associated with lone pairs, is well established.57–60 We showed a while ago that the convergent array of CH donors located close to the Co(II) ions at the fac tris-chelate cage corners, in a region of high positive electrostatic potential, provides cumulatively an H-bond donor site to guests that is comparable in strength to phenol.47 Given that phenol has been shown to be a sufficiently strong H-bond donor to form S⋯HX hydrogen bonds with CS2,57 we propose that this structure of the B·CS2 complex provides (i) a reasonable structural model for CO2 binding in the cavity,54 and (ii) a rationale for the strong preference of the cages for CO2vs. N2 binding. We note also that CS2 binds weakly in the cavity of B in aqueous solution; a standard 1H NMR titration showed that CS2 binds in fast exchange, with incremental shifts in the positions of some of the 1H NMR signals of B during addition of CS2 fitting a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm with K = 2 M−1 (ESI†).
:1 binding isotherm with K = 2 M−1 (ESI†).
The balance between absolute CO2 uptake, and CO2/N2 uptake selectivity, is amongst the best known in any kind of porous material. Fig. 4 shows data for a range of other porous materials: on this graph the results for cages A and B are clearly better than the average performance for selectivity vs. uptake for many other materials, with the results for cage B at 298 K lying furthest above the trend-line of any of the representative set of examples included in the figure.
 | ||
| Fig. 4 Logarithmic plot of ideal CO2/N2 selectivity vs. CO2 uptake capacity for a variety of porous materials. The linear trendline in black shows the general inverse relationship between gas uptake capacity and CO2/N2 selectivity. Data for the coordination cages in this work are shown as red triangles. Data for porous materials from other groups are shown for comparison as blue circles (Cooper group, ref. 32; Zhang group, ref. 34 and 35; various selected MOFs, see references within ref. 32). | ||
In conclusion we have demonstrated good CO2 uptake by a molecular cage complex in which the gaseous guest binds in the central cage cavity even when the bulk materials are not conventionally porous. Such examples of gas sorption into the cavities of molecular cages – in contrast to porous network materials – are very rare.61,62 On the basis of the structural model based on CS2, we suggest that this arises because of favourable polar interactions between the CO2 guest and charge-assisted hydrogen-bond donor sites on the interior surface of the cage host;47,53,54 these same structural features also result in particularly high selectivity for binding of CO2 compared to non-polar N2.
We thank EPSRC for financial support (grants EP/N031555/1 and EP/K503812/1).
Notes and references
- E. J. Carrington, C. A. McAnally, A. J. Fletcher, S. P. Thompson, M. Warren and L. Brammer, Nat. Chem., 2017 DOI:10.1038/Nchem.2747.
- S. A. Basnayake, J. Su, X. Zou and K. J. Balkus Jr., Inorg. Chem., 2015, 54(4), 1816–1821 CrossRef CAS PubMed.
- P. Deria, S. Li, H. Zhang, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 12478–12481 RSC.
- H. Wang, K. Yao, Z. Zhang, J. Jagiello, Q. Gong, Y. Han and J. Li, Chem. Sci., 2014, 5, 620–624 RSC.
- H. J. Jeon, R. Matsuda, P. Kanoo, H. Kajiro, L. Li, H. Sato, Y. Zheng and S. Kitagawa, Chem. Commun., 2014, 50, 10861–10863 RSC.
- M. Du, C.-P. Li, M. Chen, Z.-W. Gei, X. Wang, L. Wang and C.-S. Liu, J. Am. Chem. Soc., 2014, 136, 10906–10909 CrossRef CAS PubMed.
- M. Alhamami, H. Doan and C.-H. Cheng, Materials, 2014, 7, 3198–3250 CrossRef.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135(28), 10441–10448 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- B. Yuan, D. Ma, X. Wang, Z. Li, Y. Li, H. Liu and D. He, Chem. Commun., 2012, 48, 1135–1137 RSC.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Özgür Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2010, 1, 695–704 CrossRef PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- T. Itoh, M. Kondo, H. Sakamoto, K. Wakabayashi, M. Kanaike, K. Itami and S. Masaoka, Dalton Trans., 2015, 44, 15334–15342 RSC.
- Y. Takasaki and S. Takamizawa, J. Am. Chem. Soc., 2014, 136, 6806–6809 CrossRef CAS PubMed.
- W. Kosaka, K. Yamagashi, H. Yoshida, R. Matsuda, S. Kitagawa, M. Takata and H. Miyasaka, Chem. Commun., 2013, 49, 1594–1596 RSC.
- K. Polak-Kraśna, R. Dawson, L. T. Holyfield, C. R. Brown, A. D. Burrows and T. J. Mays, J. Mater. Sci., 2017, 52, 3862–3875 CrossRef.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Sharpe, T. C. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028–9035 CrossRef CAS PubMed.
- S. Fischer, A. Schimanowitz, R. Dawson, I. Senkovska, S. Kaskel and A. Thomas, J. Mater. Chem. A, 2014, 2, 11825–11829 CAS.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- X.-Z. Luo, X.-J. Jia., J.-H. Deng, J.-L. Zhongm, H.-J. Liu, K.-J. Wang and D.-C. Zhong, J. Am. Chem. Soc., 2013, 135(32), 11684–11687 CrossRef CAS PubMed.
- H. Ma, H. Ren, S. Meng, Z. Yan, H. Zhao, F. Sun and G. Zhu, Chem. Commun., 2013, 49, 9773–9775 RSC.
- T. Hasell, M. Miklitz, A. Stephenson, M. A. Little, S. Y. Chong, R. Clowes, L. Chen, D. Holden, G. A. Tribello, K. E. Jelfs and A. I. Cooper, J. Am. Chem. Soc., 2016, 138, 1653–1659 CrossRef CAS PubMed.
- Q. Song, S. Jiang, T. Hasell, M. Liu, S. Sun, A. K. Cheetham, E. Sivaniah and A. I. Cooper, Adv. Mater., 2016, 28, 2629–2637 CrossRef CAS PubMed.
- R. Manurung, D. Holden, M. Miklitz, L. Chen, T. Hasell, S. Y. Chong, M. Harancyzk, A. I. Cooper and K. E. Jelfs, J. Phys. Chem. C, 2015, 119, 22577–22586 CAS.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- D. Holden, K. E. Jelfs, A. Trewin, D. J. Willock, M. Harancyzk and A. I. Cooper, J. Phys. Chem. C, 2014, 118, 12734–12743 CAS.
- S. Jiang, K. E. Jelfs, D. Holden, T. Hasell, S. Y. Chong, M. Haranczyk, A. Trewin and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 17818–17830 CrossRef CAS PubMed.
- T. Hasell, J. A. Armstrong, K. E. Jelfs, F. H. Tay, K. M. Thomas, S. G. Kazarian and A. I. Cooper, Chem. Commun., 2013, 49, 9410–9412 RSC.
- S. Jiang, J. Bacsa, X. Wu, J. T. A. Jones, R. Dawson, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Commun., 2011, 47, 8919–8921 RSC.
- T. Mitra, X. Wu, R. Clowes, J. T. A. Jones, K. E. Jelfs, D. J. Adams, A. Trewin, J. Basca, A. Steiner and A. I. Cooper, Chem. – Eur. J., 2011, 17, 10235–10240 CrossRef CAS PubMed.
- Y. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650–6658 CrossRef CAS PubMed.
- Y. Jin, B. A. Voss, R. D. Noble and W. Zhang, Angew. Chem., Int. Ed., 2010, 49(36), 6348–6351 CrossRef CAS PubMed.
- J. Tian, J. Liu, J. Liu and P. K. Thallapally, CrystEngComm, 2013, 15, 1528–1531 RSC.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 128, 698–699 CrossRef PubMed.
- P. K. Thallapally, G. O. Lloyd, T. B. Wirsig, M. W. Bredenkamp, J. L. Atwood and L. J. Barbour, Chem. Commun., 2005, 5272–5274 RSC.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816–1820 CrossRef CAS PubMed.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 127, 13134–13135 CrossRef PubMed.
- J. L. Atwood, L. J. Barbour and A. Jerga, Angew. Chem., Int. Ed., 2004, 43, 2948–2950 CrossRef CAS PubMed.
- J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- S. D. Burd, S. Ma, J. A. Perman, B. J. Sikora, R. Q. Snurr, P. K. Thallapally, J. Tian, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 3663–3666 CrossRef CAS PubMed.
- S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. López-Garzón, M. Domingo-García, I. Fernández-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664–1672 CrossRef CAS.
- M. D. Ward, C. A. Hunter and N. H. Williams, Chem. Lett., 2017, 46, 2–9 CrossRef.
- S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122–1132 CrossRef CAS PubMed.
- I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167–15175 CrossRef CAS PubMed.
- M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744–2751 RSC.
- S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475–8483 CrossRef CAS PubMed.
- W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 2790–2794 RSC.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- A. J. Metherell and M. D. Ward, Dalton Trans., 2016, 45, 16096–16111 RSC.
- C. G. Morris, N. M. Jacques, H. G. W. Godfrey, T. Mitra, D. Fritsch, Z. Lu, C. A. Murray, J. Potter, T. M. Cobb, F. Yuan, C. C. Tang, S. Yang and M. Schröder, Chem. Sci., 2017, 8, 3239–3248 RSC.
- M. R. Battaglia, A. D. Buckingham, D. Neumark, R. K. Pierens and J. H. Williams, Mol. Phys., 1981, 43, 1015–1020 CrossRef CAS.
- G. L. D. Ritchie and J. Vrbancich, J. Chem. Soc., Faraday Trans. 2, 1980, 76, 1245–1248 RSC.
- A. B. Sannigrahi and A. K. Chandra, Bull. Chem. Soc. Jpn., 1967, 40, 1344 CrossRef CAS.
- A. L. Picone and R. M. Romano, J. Mol. Struct., 2010, 978, 187 CrossRef CAS.
- M. Wierzejewska and M. Dziadosz, J. Mol. Struct., 1999, 513, 155 CrossRef.
- K. N. Power, T. L. Hennigar and M. J. Zaworotko, New J. Chem., 1998, 177–181 RSC.
- I. A. Riddell, M. M. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC.
- J. Roukala, J. Zhu, C. Giri, K. Rissanen, P. Lantto and V.-V. Telkki, J. Am. Chem. Soc., 2015, 137, 2464–2467 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the crystal structure of [Co8(Lw)12](BF4)16·CS2·5H2O; powder XRD analyses; gas sorption data at 273 K; NMR titration data for binding of CS2 into B in aqueous solution; additional figures. CCDC 1537994. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc01959a |
| ‡ Equal first authors. |
| This journal is © The Royal Society of Chemistry 2017 |