
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Somatostatin receptor-targeted organometallic iridium(III) complexes as novel theranostic agents†
Vojtech
Novohradsky
a,
Ana
Zamora
bc,
Albert
Gandioso
b,
Viktor
Brabec
 *a,
José
Ruiz
*c and
Vicente
Marchán
*b
*a,
José
Ruiz
*c and
Vicente
Marchán
*b
aInstitute of Biophysics, Academy of Sciences of the Czech Republic, v.v.i. Kralovopolska 135, 612 65 Brno, Czech Republic. E-mail: brabec@ibp.cz
bDepartament de Química Inorgànica i Orgànica, Secció de Química Orgànica, IBUB, Universitat de Barcelona, E-08028 Barcelona, Spain. E-mail: vmarchan@ub.edu
cDepartamento de Química Inorgánica, Universidad de Murcia, and Institute for Bio-Health Research of Murcia (IMIB-Arrixaca), E-30071 Murcia, Spain. E-mail: jruiz@um.es
First published on 25th April 2017
Abstract
A novel somatostatin receptor-targeted anticancer agent based on the conjugation of a highly cytotoxic and luminescent cyclometalated iridium(III) complex to tumor-targeting vectors based on octreotide peptide has been described, and its potential for targeted theranostic applications has been demonstrated.
During the fight against cancer, chemotherapeutic agents have to overcome many obstacles that often prevent a successful outcome of the disease. Indeed, low molecular weight cytotoxic drugs, either organic molecules (e.g. camptothecin and doxorubicin) or metal complexes (e.g. cisplatin and derivatives), cause severe toxic side effects in patients owing to their poor tumor tissue selectivity. Low tumor accumulation, poor aqueous solubility and intrinsic or acquired resistance also contribute to reducing their anticancer efficacy. In such a context, targeted delivery approaches1 have emerged as a promising strategy to overcome these drawbacks, particularly those based on ligands whose receptors are overexpressed on the surface of malignant cells compared with healthy cells.1,2 The conjugation of therapeutic agents to targeting vehicles based on small regulatory peptides offers several advantages including the disposal of efficient solid-phase procedures for synthesizing drug conjugates with improved pharmacological properties.
Iridium complexes have recently emerged as promising alternatives to platinum-based metallo-anticancer drugs.3 Meanwhile, cyclometalated Ir(III) complexes have gained attention as imaging and sensing probes due to their rich photophysical properties and good cell permeability,4 which can be fine-tuned by the modification of the ligands. For example, the use of the pharmacophore benzimidazole as a ligand has given rise to a wide variety of metal compounds that act either as anti-angiogenic and/or anti-tumor agents,5 or inhibitors of amyloid-β aggregation.6 The integration of anticancer activities into cyclometalated Ir(III) complexes provides, therefore, an opportunity for the construction of novel theranostic platforms. Ir(III) complexes can also function as efficient photosensitizers for producing singlet oxygen, 1O2, and can even be developed as organelle-targeted PDT agents.7 Despite these promising achievements, further work is necessary to improve the pharmacological properties of Ir(III) metallodrugs, such as aqueous solubility and selectivity against cancer cells. In this context, targeted approaches based on peptide vectors whose receptors are overexpressed on the membrane of tumor cells compared with normal cells2,8 in combination with light activation open the door to a new generation of metallo-anticancer agents with a dual mechanism of selectivity.9
Herein, we have conjugated for the first time a highly cytotoxic and luminescent Ir(III) complex, [Ir(ppz)2(N∧N)] (Hppz = 1-phenylpyrazole; N∧N = methyl 1-butyl-2-pyridyl-benzimidazole-5-carboxylate),5a to tumor-targeting vectors based on octreotide peptide with the aim of increasing cancer cell selectivity and exploring their potential as novel theranostic agents (Scheme 1).
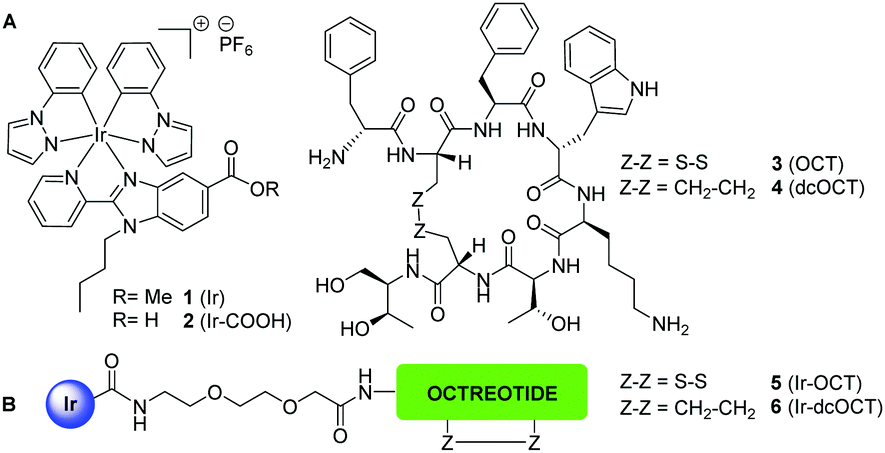 | ||
| Scheme 1 (A) Structure of the cyclometalated Ir complexes (1 and 2) and octreotide (3) and its dicarba analogue (4). (B) Schematic representation of the Ir–octreotide conjugates (5 and 6). | ||
Octreotide (OCT) is a FDA-approved synthetic cyclooctapeptide agonist of the endocrine hormone somatostatin that displays high affinity for the somatostatin subtype-2 receptor (SSTR2).2a It is metabolically more stable than somatostatin since it incorporates D-amino acids, and the cysteine bridge stabilizes the pharmacophore sequence (Phe7-D-Trp8-Lys9-Thr10) in a β-turn. As a result, octreotide binds with high affinity and selectivity to somatostatin receptors (SSTRs), mainly SSTR2.2a,10 Precisely, the fact that SSTR2 is the most frequently overexpressed somatostatin receptor on the membrane of many tumor cells led to the use in the clinics of the [111In-DTPA]-octreotide and [90Y-DOTA-Tyr3]-octreotide conjugates in molecular imaging and therapy of neuroendocrine tumors, respectively,2a,10 and several other SSTR2-targeted radiotherapeutics are currently under clinical evaluation.1a Octreotide has also been conjugated to cytotoxic organic drugs and metal complexes, with promising results because of the reduced toxicity and the increased selectivity.11
The attachment of the Ir(III) complex to octreotide was designed through the formation of an amide bond between a carboxylic function in the benzimidazole diimine ligand and the N-terminal end of the peptide sequence (Scheme 1). The incorporation of a spacer is essential to keep the metal complex away from the pharmacophore sequence and the β-turn peptide structure, which are key elements for recognition and binding to the receptor.11b Besides octreotide, we also selected a dicarba analogue (dcOCT), since the replacement of the disulfide bond by a CH2–CH2 linkage increases stability in the reductive cellular environment without significantly altering the binding affinity for somatostatin receptors.11b,c,12
First, the ester function of the parent cyclometalated Ir(III) complex 15a was hydrolyzed with LiOH to afford complex 2 (Scheme 1) bearing the carboxylic function suitable for conjugation. Taking into consideration the advantages of solid-phase peptide synthesis (SPPS), the synthesis of the Ir–OCT (5) and Ir–dcOCT (6) conjugates was planned through a stepwise solid-phase strategy in which the metallic moiety is regioselectively attached at the N-terminal end of the resin-bound peptide (Scheme 2). In both cases, the linear peptide sequences were assembled manually on a Rink amide resin-p-MBHA using standard Fmoc-tBu methodology (Scheme 2).11b,c In the case of 5, after the attachment of 2, side-chain deprotection and cleavage from the resin afforded the linear conjugate, which was cyclized via disulfide bond formation. On the other hand, once the linear dicarba analogue of octreotide, where both cysteines are replaced by allyl glycine residues, had been assembled, on-resin microwave-assisted ring-closing metathesis with a second-generation Grubbs catalyst followed by hydrogenation using Wilkinson's catalyst afforded the protected dicarba analogue of octreotide bound to the resin (Scheme 2).11b,c Finally, after the incorporation of 2, acidic treatment afforded directly 6. Both conjugates were purified by reversed-phase HPLC and fully characterized by HR ESI-MS, UV-vis and fluorescence spectroscopy (Fig. S3–S8, ESI†). Importantly, the aqueous solubility of 1 was substantially improved upon peptide derivatization since both conjugates were found to be completely soluble in PBS buffer.
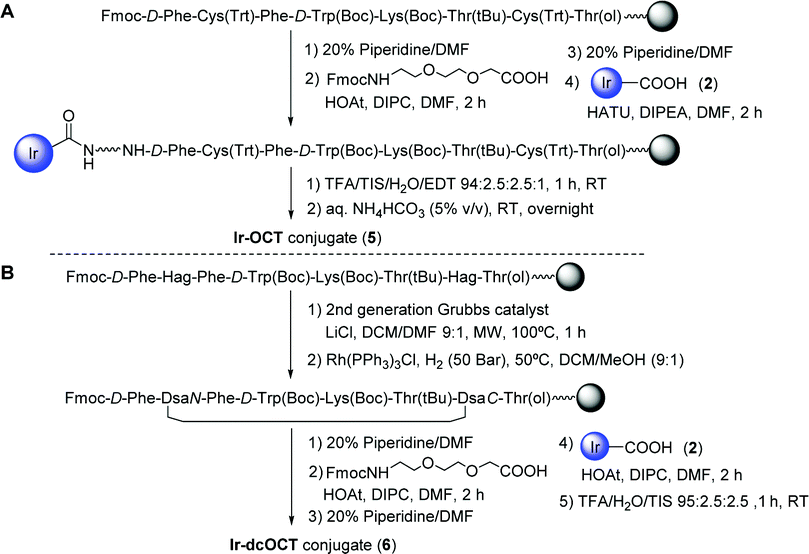 | ||
| Scheme 2 Schematic representation of the solid-phase approach used for the synthesis of the Ir–OCT (5) (A) and Ir–dcOCT (6) (B) conjugates. | ||
Having at hand Ir–octreotide conjugates, we first studied the expression levels of the hSSTR2 receptor in a panel of cancer cell lines to choose the best model for assessing the capacity of the peptides to deliver the Ir(III) drug in a selective manner. As shown in Fig. S9 and Table S1 (ESI†), HeLa cervix carcinoma cells expressed high levels of hSSTR2 while MDA-MB-231 breast cancer cells expressed low levels and, therefore, they can be used as positive and negative controls of cells over-expressing SSTR2, respectively. Next, the efficiency of intracellular delivery of octreotide, OCT, and of the dicarba analogue, dcOCT, was studied in HeLa cells by flow cytometry by using the corresponding fluorescein-labeled peptides.11b As previously found by us in MCF-7 cells,11b the internalization of FITC-OCT was slightly higher compared with that of FITC-dcOCT, which correlates with the lower binding affinity of dcOCT for SSTR2 compared with OCT (Fig. S10, ESI†).10a,12
Since the cytotoxic activity of metal-based anticancer drugs is highly dependent on cellular uptake and accumulation, we studied the effect of peptide conjugation on the accumulation of the Ir(III) complex as well as the participation of SSTR2 in this process. For this purpose, the accumulation of the Ir–octreotide conjugates (5 and 6) was studied in HeLa and MDA-MB-231 cancer cells and compared with that of the parent complexes (1 and 2). Iridium accumulation was quantitatively determined by ICP-MS after a 2 h exposure treatment to 5 μM of the compounds at 37 °C (Tables S2 and S3, ESI†). As shown in Fig. 1, complex 1 accumulates much better than the rest of the compounds in both cell lines, which can be attributed both to its high hydrophobicity and to its cationic nature.5a By contrast, the accumulation of complex 2 bearing the carboxylic function was almost negligible in both cell lines. Although Ir–octreotide conjugates accumulate to a lesser extent than 1, in both cases the accumulation was slightly higher in HeLa (SSTR2+) than in MDA-MB-231 (SSTR2−) cells. As expected from the low binding affinity of dcOCT for SSTR2 compared with OCT,12 conjugate 5′ accumulation was higher than that of conjugate 6. In order to get more insights into the cellular uptake of the compounds, we repeated Ir accumulation studies at 4 °C. As previously described for other Ir(III) complexes,4d,7b the incubation of HeLa and MDA-MB-231 cells with 1 at low temperature led to a reduction in the Ir accumulation, which indicates that 1 enters the cells through an energy-dependent pathway and not exclusively by passive diffusion. Very interestingly, the accumulation of 5 and 6 was reduced in SSTR2+ HeLa cells at 4 °C, thereby indicating internalization through an SSTR2-mediated energy-dependent endocytic mechanism. However, the accumulation of the Ir–octreotide conjugates in the SSTR2− MDA-MB-231 cells was not influenced when lowering the temperature, which could suggest the participation of other penetration mechanisms. These results are not surprising since the nature of the targeted drug and of the linker can strongly influence not only peptide–receptor binding but also tumor penetration, metabolism and excretion of the conjugate.1a
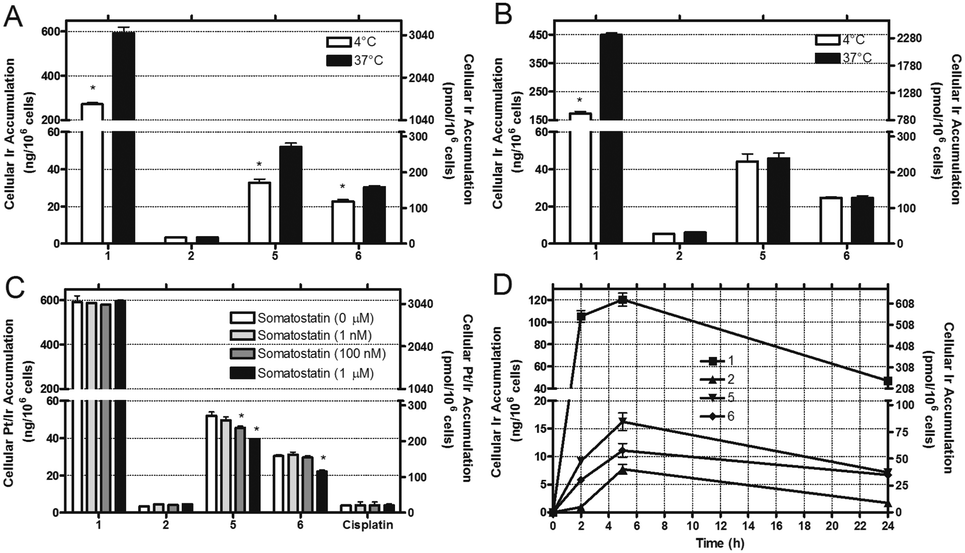 | ||
| Fig. 1 Accumulation studies of Ir compounds (5 μM) determined by ICP-MS in HeLa (A) and MDA-MB-231 (B) cells after treatment for 2 h at 37 °C or 4 °C. (C) Accumulation of Ir compounds and cisplatin (5 μM, 2 h, 37 °C) determined by ICP-MS in HeLa cells. Cells were first pre-treated for 1 h with somatostatin at increasing concentrations. (D) Kinetics of cellular accumulation of Ir compounds in HeLa cells after incubation at 1 μM for the indicated time period at 37 °C. Results are the mean ± SDs from three independent experiments. | ||
Taking into account the above-mentioned results, we carried out competitive studies with somatostatin to further confirm the involvement of SSTR2 in the internalization of the conjugates. As shown in Fig. 1C, the pre-treatment of HeLa cells with somatostatin led to a concentration-dependent reduction of the accumulation of both conjugates, which confirms the participation of SSTR2. As expected, neither Ir accumulation from complex 1 or 2 nor Pt accumulation from cisplatin was affected by the presence of somatostatin. Interestingly, accumulation in HeLa cells at different time periods revealed a significant drop in the total cellular Ir accumulation after 24 h, which suggests that the Ir compounds or their metabolites are good substrates for p-glycoprotein or some other alternative detoxification mechanisms (Fig. 1D).
Cytotoxicity studies were performed in HeLa and MDA-MB-231 cells to estimate the in vitro antitumor potential of Ir–octreotide conjugates (5 and 6) and of the parent complexes (1 and 2) and peptides (OCT and dcOCT). The photobiological activity of the compounds was also assessed via irradiation with visible light. In both cases, the MTT assay was performed after 24 h (Table 1) or 72 h (Table S4, ESI†). Not surprisingly, complex 1 displayed the highest activity under all the tested conditions,5a and visible light irradiation leads to a slight improvement of the IC50. In contrast, complex 2 did not display antiproliferative activity. These results correlate well with the accumulation data of both complexes determined by ICP-MS. The conjugation of the Ir(III) complex to the peptide moieties reduced the cytotoxic activity of 1 in both cell lines, which again correlates with the reduced accumulation. However, the activity that the conjugates retain is still reasonable for a drug–peptide conjugate, specially taking into account that their efficacy depends not only on the potency of the therapeutic cargo but also on several factors such as the number of receptors available to mediate internalization, the receptor recycling rate, the binding affinity of the peptide for its receptor when conjugated to the drug cargo or endosomal sequestration, among others.1a Overall, both conjugates showed similar antitumor activities which attest the CH2–CH2 linkage as a suitable isostere for the disulfide bond. Even so, Ir–OCT was about 2–3 times more active than Ir–dcOCT and was capable of distinguishing between HeLa and MDA-MB-231 cells after 24 h, being more active in the SSTR2-overexpressing cell line. This is particularly appealing since the parent complex 1 was more active (about 2.5-fold) in the SSTR2− MDA-MB-231 cells. Similarly to complex 1, visible light irradiation drives to a moderate improvement of the antitumor activities of the conjugates, enough however to balance the activity reduction that conjugation involves. Interestingly, the cytotoxic activity of conjugate 5 after 24 h was slightly lower in the non-malignant CHO-K1 cell line than in tumor HeLa cells (Table S4, ESI†). Octahedral Ir(III) complexes are characterized for being highly photostable, so that they are rarely used in PACT7a while the generation of reactive oxygen species (ROS) is the main mechanism for their PDT-initiated cell death. Therefore, the intracellular production of ROS was studied in HeLa cells, either in the dark or after visible light irradiation. As shown in Fig. S11 (ESI†), the cellular ROS generation was higher after light irradiation, which suggests that disruption of the mitochondrial function could also be involved in their mechanism of action. As expected, ROS production from the parent complex 1 was higher than that of the Ir–octreotide conjugates, which again correlates with higher accumulation.
| HeLa | MDA-MB-231 | |||||
|---|---|---|---|---|---|---|
| Dark | Irradiated | PIb | Dark | Irradiated | PIb | |
| a Results are the means ± SDs from three independent experiments. b PI: phototoxic index (IC50 of non-irradiated cells/IC50 irradiated cells). | ||||||
| 1 | 3.13 ± 0.21 | 1.25 ± 0.11 | 2.5 | 1.23 ± 0.09 | 0.91 ± 0.08 | 1.4 |
| 2 | >200 | >200 | nd | >200 | >200 | nd |
| 5 | 30.9 ± 2.7 | 15.5 ± 2.5 | 2.0 | 49.2 ± 4.1 | 41.9 ± 4.4 | 1.2 |
| 6 | 57.8 ± 4.9 | 53.1 ± 5.1 | 1.1 | 51.0 ± 3.3 | 36.7 ± 2.8 | 1.4 |
| OCT | >200 | >200 | nd | >200 | >200 | nd |
| dcOCT | >200 | >200 | nd | >200 | >200 | nd |
Conjugates between targeting ligands and highly cytotoxic luminescent complexes are attractive candidates for the development of novel theranostic agents13 combining within one molecular system a potent cytotoxic effect with cell targeting and imaging capabilities. In this context, we have recently found that Ir(III) complexes of the type 1 mainly accumulate in the cytoplasm, specifically in the actin cortex.5a Based on these precedents, the cellular localization of the Ir–octreotide conjugates was studied by laser scanning confocal microscopy in HeLa cells with the aim of exploring their potential as targeted theranostic agents. As shown in Fig. 2 and Fig. S12 (ESI†), the emission of the Ir(III) complex allowed the visualization of luminescent vesicles in the cytoplasm, most likely endosomes, confirming the cellular uptake of the Ir–octreotide conjugates by HeLa cells. Slightly lower cellular accumulation in MDA-MB-231 cells was reflected from the reduced intensity of the luminescence signal compared to that of HeLa cells (Fig. S13, ESI†).
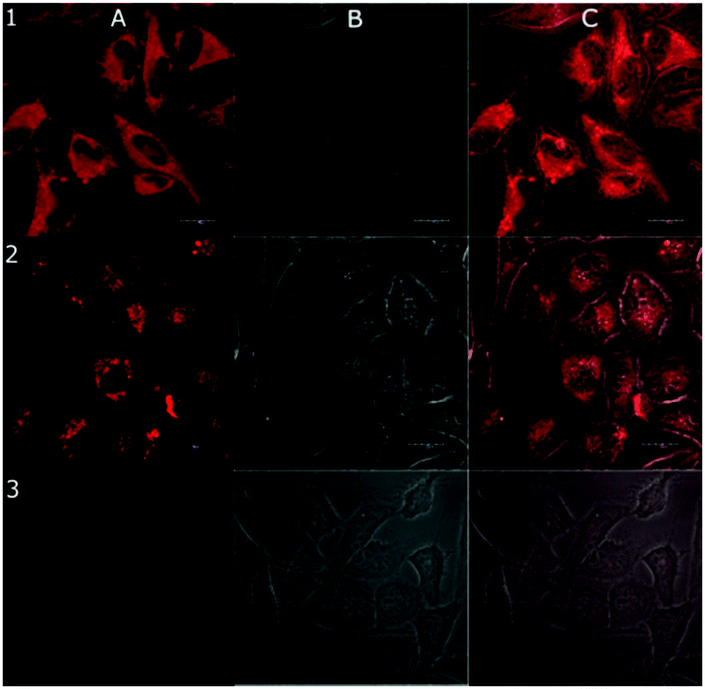 | ||
| Fig. 2 Confocal microphotograph of HeLa cells treated for 24 h with 1 μM of 1 (1), 5 (2), or untreated control cells (3). Luminescent channel (A), bright field (B) and (C) merged images. Scale bars: 20 μm. | ||
In summary, we have described the synthesis of novel somatostatin-targeted anticancer agents based on the conjugation of a cyclometalated luminescent Ir(III) complex to octreotide vehicles, and demonstrated their potential as targeted theranostic agents. On the one hand, Ir–octreotide conjugates accumulate in cancer cells overexpressing SSTR2, and the participation of the receptor was confirmed by competitive experiments. Such differences in accumulation between SSTR2+ and SSTR2− cancer cell lines allowed the modification of the cytotoxicity of the parent Ir complex, since the conjugates were more active in HeLa cells than in MDA-MB-231 cells, which is the opposite tendency found with 1. Notably, peptide vehicles (OCT and dcOCT) were non-cytotoxic and the cytotoxicity was increased in all cases upon visible light irradiation and ROS production was confirmed. On the other hand, the internalization of the Ir–octreotide conjugates could be easily visualized by confocal microscopy owing to the luminescence properties of the Ir(III) complex. Overall, these results open up the door to the design of novel theranostic agents based on Ir–peptide conjugates with improved tumor selectivity. Future work is directed to the optimization of the compounds by improving the potency and photophysical properties of the cyclometalated Ir(III) complex through ligand modifications5a as well as by exploring the use of more hydrophilic or cleavable linkers to improve their pharmacological properties.1a
This work was supported by the Spanish Ministry of Economy and Competitiveness and FEDER funds (Projects CTQ2015-64319-R and CTQ2014-52658-R), the Czech Science Foundation (Grant 17-09436S), COST CM1105, CM1406 and the MetDrugs network (CTQ2015-70371-REDT). A. Z. thanks Fundacion Séneca-CARM for a grant (Exp. 19020/FPI/13).
Notes and references
- (a) M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203 CrossRef CAS PubMed; (b) G. Trapani, N. Denora, A. Trapani and V. Laquintana, J. Drug Targeting, 2012, 20, 1 CrossRef PubMed.
- (a) G. Mezo and M. Manea, Expert Opin. Drug Delivery, 2010, 7, 79 CrossRef CAS PubMed; (b) F. Danhier, A. Le Breton and V. Préat, Mol. Pharmaceutics, 2012, 9, 2961 CrossRef CAS PubMed.
- (a) Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174 CrossRef CAS PubMed; (b) Z. Liu, I. Romero-Canelon, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., Int. Ed., 2014, 53, 3941 CrossRef CAS PubMed; (c) A. Kastl, A. Wilbuer, A. L. Merkel, L. Feng, P. Di Fazio, M. Ocker and E. Meggers, Chem. Commun., 2012, 48, 1863 RSC; (d) N. Cutillas, G. S. Yellol, C. de Haro, C. Vicente, V. Rodríguez and J. Ruiz, Coord. Chem. Rev., 2013, 257, 2784 CrossRef CAS; (e) J. Yellol, S. A. Pérez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, J. Med. Chem., 2015, 58, 7310 CrossRef CAS PubMed.
- (a) T. S.-M. Tang, K.-K. Leung, M.-W. Louie, H.-W. Liu, S. H. Cheng and K. K.-W. Lo, Dalton Trans., 2015, 44, 4945 RSC; (b) D.-L. Ma, D. S.-H. Chan and C.-H. Leung, Acc. Chem. Res., 2014, 47, 3614 CrossRef CAS PubMed; (c) X. Li, X. Tong, H. Yan, C. Lu, Q. Zhao and W. Huang, Chem. – Eur. J., 2016, 22, 17282 CrossRef CAS PubMed; (d) C. Li, Y. Liu, Y. Wu, Y. Sun and F. Li, Biomaterials, 2013, 34, 1223 CrossRef CAS PubMed.
- (a) J. Yellol, S. A. Perez, G. Yellol, J. Zajac, A. Donaire, G. Vigueras, V. Novohradsky, C. Janiak, V. Brabec and J. Ruiz, Chem. Commun., 2016, 52, 14165 RSC; (b) G. S. Yellol, A. Donaire, J. G. Yellol, V. Vasylyeva, C. Janiak and J. Ruiz, Chem. Commun., 2013, 49, 11533 RSC.
- G. S. Yellol, J. G. Yellol, V. B. Kenche, X. M. Liu, K. J. Barnham, A. Donaire, C. Janiak and J. Ruiz, Inorg. Chem., 2015, 54, 470 CrossRef CAS PubMed.
- (a) K. K.-S. Tso, K.-K. Leung, H.-W. Liu and K. K.-W. Lo, Chem. Commun., 2016, 52, 4557 RSC; (b) J.-J. Cao, C.-P. Tan, M. H. Chen, N. Wu, D.-Y. Yao, X.-G. Liu, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2017, 8, 631 RSC.
- M. Soler, L. Feliu, M. Planas, X. Ribas and M. Costas, Dalton Trans., 2016, 45, 12970 RSC.
- (a) A. Gandioso, E. Shaili, A. Massaguer, G. Artigas, A. Gonzalez-Cantó, J. A. Woods, P. J. Sadler and V. Marchán, Chem. Commun., 2015, 51, 9169 RSC; (b) A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044 RSC; (c) F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098 CrossRef PubMed; (d) T. Wang, N. Zabarska, Y. Wu, M. Lamla, S. Fischer, K. Monczak, D. Y. W. Ng, S. Rau and T. Weil, Chem. Commun., 2015, 51, 12552 RSC.
- (a) A. Janecka, M. Zubrzycka and T. Janecki, J. Pept. Res., 2001, 58, 91 CrossRef CAS PubMed; (b) J. C. Reubi, Endocr. Rev., 2003, 24, 389 CrossRef CAS PubMed.
- (a) L.-C. Sun and D. H. Coy, Drugs Future, 2008, 33, 217 CrossRef CAS; (b) F. Barragán, D. Carrion-Salip, I. Gómez-Pinto, A. González-Cantó, P. J. Sadler, R. de Llorens, V. Moreno, C. González, A. Massaguer and V. Marchán, Bioconjugate Chem., 2012, 23, 1838 CrossRef PubMed; (c) F. Barragan, V. Moreno and V. Marchán, Chem. Commun., 2009, 4705 RSC.
- D. D'Addona, A. Carotenuto, E. Novellino, V. Piccand, J. C. Reubi, A. Di Cianni, F. Gori, A. M. Papini and M. Ginanneschi, J. Med. Chem., 2008, 51, 512 CrossRef PubMed.
- (a) R. Kumar, W. Sup Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670 RSC; (b) B. Bertrand, P.-E. Doulain, C. Goze and E. Bodio, Dalton Trans., 2016, 45, 13005 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization and biological evaluation of the compounds. See DOI: 10.1039/c7cc01946g |
| This journal is © The Royal Society of Chemistry 2017 |