
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P-Stereogenic bisphosphines with a hydrazine backbone: from N–N atropoisomerism to double nitrogen inversion†
Amparo
Prades
 a,
Samuel
Núñez-Pertíñez
a,
Antoni
Riera
*ab and
Xavier
Verdaguer
*ab
a,
Samuel
Núñez-Pertíñez
a,
Antoni
Riera
*ab and
Xavier
Verdaguer
*ab
aInstitute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology, Baldiri Reixac 10, E-08028, Spain. E-mail: antoni.riera@irbbarcelona.org; xavier.verdaguer@irbbarcelona.org; Tel: +34 934034813
bDept. Química Inorgànica i Orgànica, Secció de Química Orgànica, Universitat de Barcelona, Martí i Franquès 1, E-08028 Barcelona, Spain
First published on 31st March 2017
Abstract
The synthesis of P-stereogenic bisphosphine ligands starting from a phosphinous acid chiral synthon and hydrazine is reported. The dialkylation of the hydrazine backbone yielded atropo- and nitrogen inversion isomers which are in slow exchange. The crystallization of one of the isomers allowed us to study the reaction kinetics of the equilibria. The new ligands were tested in the Rh catalysed asymmetric hydrogenation of various benchmark substrates attaining up to 99% ee.
Developing active and selective chiral catalysts is highly relevant for the chemical and pharmaceutical industries.1 “Ligand engineering” is a key approach to generate new and more efficient catalytic systems. Among the different types of ligand, chiral phosphines have made a key contribution to asymmetric catalysis, which is one of the most effective synthetic methodologies by which to obtain optically pure compounds.2 Since the pioneering work of Kagan with DIOP3 and Knowles with DIPAMP,4 a large number of chiral bisphosphine ligands have been reported. Among these, P-stereogenic phosphines have proved to be a privileged class of ligands.5,6 Despite enormous advances in this field, P-stereogenic ligands are usually made through a lengthy multistep synthesis. Thus, the search for cost efficient synthesis of optically pure P-stereogenic ligands is of key importance.
Our group has recently reported procedures for the synthesis of optically pure P-stereogenic tert-butylmethylphosphinous acid borane 1.7 This fragment is highly versatile and has allowed access to C1 symmetric MaxPHOS8 and MaxPHOX9 ligands. In this context, we addressed whether our methodology could also be amenable for the rapid and efficient synthesis of ligands with C2 symmetry. We hypothesized that the coupling of hydrazine to the activated phosphinous acid 1 could provide nitrogen analogs of bis-P* type ligands (Scheme 1).10 Here we report the synthesis of two borane-protected P-stereogenic hydrazine bisphosphine ligands derived from hydrazine and 1. In solution, these ligands are present as a mixture of atropo- and nitrogen inversion isomers in slow exchange. These ligands were also tested in the Rh catalyzed asymmetric hydrogenation, attaining selectivities up to 99% ee.
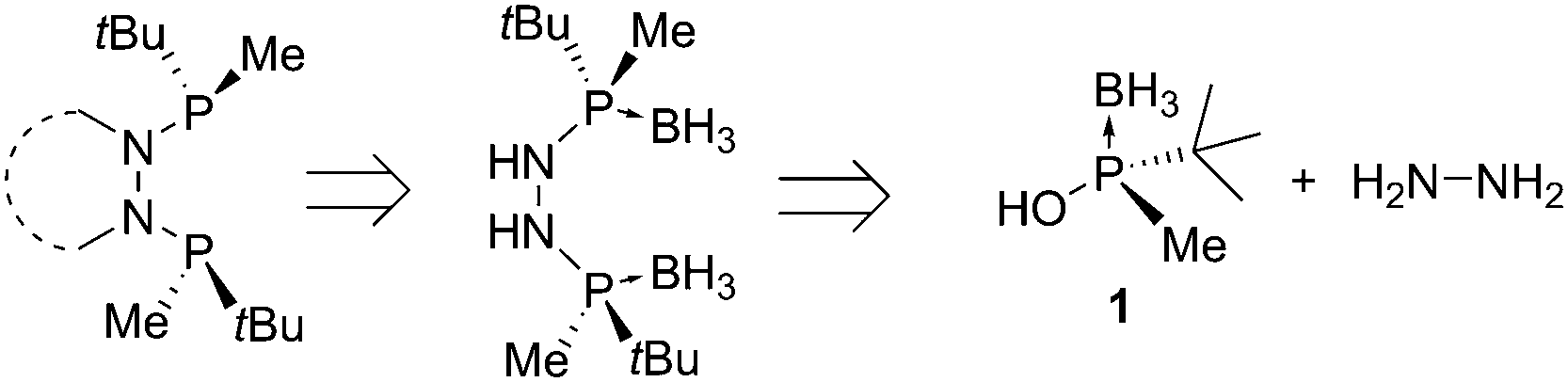 | ||
| Scheme 1 Strategy developed for the synthesis of P-stereogenic C2 bisphosphines with a hydrazine backbone. | ||
Initially, in order to identify all possible intermediates, a sequential synthesis was performed, starting from 1 and Boc-hydrazine (Scheme 2). Phosphinous acid 1 was activated with Ms2O following our procedure.7c The reaction with tert-butyl carbazate at −20 °C provided hydrazine 2 (58%) in an SN2@P-type process. The reaction was stereospecific and occurred with inversion of configuration at the phosphorus center. The Boc protecting group was removed using anhydrous HCl in MeOH to yield phosphino hydrazine 3 in 72% yield. From 3, a second phosphinous acid coupling provided bisphosphine 4 in 56% yield. Satisfactory stereocontrol was observed throughout the syntheses, and meso-4 was not detected. We next attempted the one-pot preparation of 4 starting from hydrazine and 2 equivalents of phosphinous acid 1. We observed that 4 was assembled efficiently in one step with 82% yield.
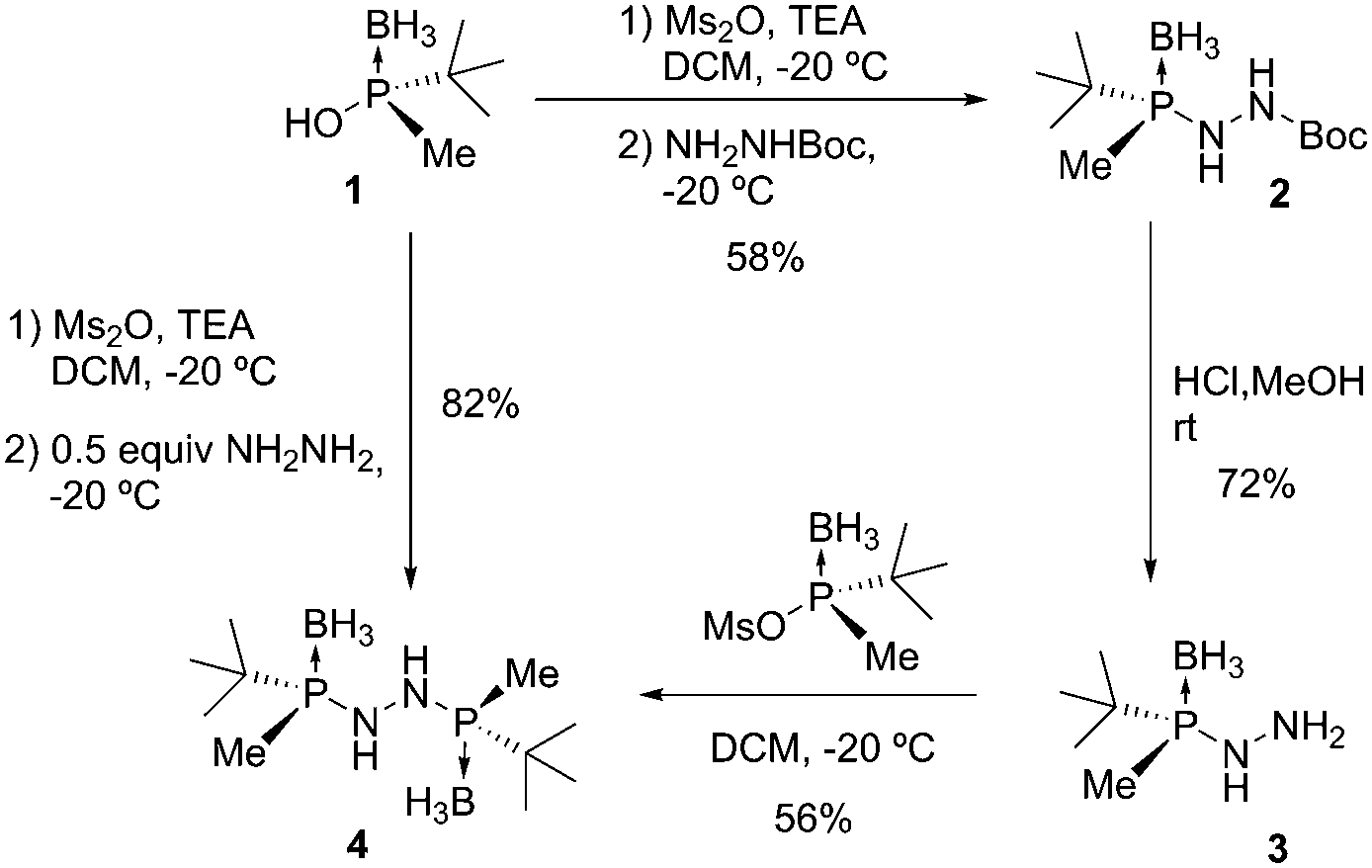 | ||
| Scheme 2 Sequential and direct synthesis of the C2 bisphosphine hydrazine 4. | ||
To improve the stability of the resulting ligands, we derivatized 4 by alkylation on both nitrogen atoms.11 The methylation of 4 was achieved by deprotonating hydrazine nitrogen atoms with sodium hydride and adding an excess of methyl iodide at 55 °C (Scheme 3). The dimethyl derivative 5 was isolated in 90% yield. When allyl bromide was used as the alkylating agent, only monoalkylated product 6 was obtained in moderate yield (49%). The dialkylated product was not observed, even when the reaction temperature was increased and the solvent changed. At this stage, a large number of electrophiles were also tested; however, dialkylation products were not detected. A cyclization reaction was attempted with 1,3-dibromopropane as the alkylating agent. However, the reaction did not afford the expected pyrazolidine ring. The NMR spectra were in concordance with the formation of 6, which results from the monoalkylation of 4 followed by base-promoted elimination of HBr. Finally, we deduced that, by using 3-bromo-2-bromomethyl-1-propene, we could avoid the undesired E2 type reaction. Indeed, using this electrophile, the cyclization occurred swiftly, providing the corresponding pyrazolidine 7 in 77% yield.
 | ||
| Scheme 3 N-Alkylation of the hydrazine backbone. | ||
1H NMR showed that crystalline 5 was present as a single compound; however, upon dissolution, it rapidly equilibrated to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of isomers. Analysis of 5 by thin-layer chromatography (TLC) on silica provided two spots with retention factors of 0.3 and 0.5 (hexane:AcOEt 8:2). However, when the reaction crude product was eluted through a SiO2 flash column, the two substances were equally present in all the fractions. 2D-TLC analysis showed that, even in a quick elution, the two products could not be isolated. The system re-equilibrated a second time, with a new set of spots on the TLC appearing for every single spot in the first elution. This observation suggested that 5 was present as a mixture of two isomers in slow exchange. A variable temperature 1H-NMR experiment of a solution of 5 in DMSO-d6 was performed at 25, 40 and 70 °C. During the experiment, the signals corresponding to the protons of the NCH3 groups of the two species got closer, but total coalescence was not observed, and at 100 °C the sample decomposed.
:1 mixture of isomers. Analysis of 5 by thin-layer chromatography (TLC) on silica provided two spots with retention factors of 0.3 and 0.5 (hexane:AcOEt 8:2). However, when the reaction crude product was eluted through a SiO2 flash column, the two substances were equally present in all the fractions. 2D-TLC analysis showed that, even in a quick elution, the two products could not be isolated. The system re-equilibrated a second time, with a new set of spots on the TLC appearing for every single spot in the first elution. This observation suggested that 5 was present as a mixture of two isomers in slow exchange. A variable temperature 1H-NMR experiment of a solution of 5 in DMSO-d6 was performed at 25, 40 and 70 °C. During the experiment, the signals corresponding to the protons of the NCH3 groups of the two species got closer, but total coalescence was not observed, and at 100 °C the sample decomposed.
The X-ray structure of 5 is depicted in Fig. 1.12 The sum of the angles around the trisubstituted nitrogen atoms added up to 360.0 and 359.9°, which indicates that the N atoms were completely planar. The view through the N–N axis shows how the two N planes are almost perpendicular, the P–N–N–P torsion angle being 98.0°. All these findings sustain that the two species observed in solution are atropoisomers that arise from the hampered rotation around the N–N bond caused by the increased steric hindrance of the phosphorus groups (Fig. 1c).13 Because of the presence of P-stereogenic centers, the new axial chirality leads to two distinct diastereoisomers (RP,RP,Ra)-5 and (RP,RP,Sa)-5, which can be observed as two sets of signals in the 1H NMR spectrum. As (RP,RP,Sa)-5 was the only atropoisomer present in the solid state, a kinetics study was performed to determine the activation energy of the equilibrium between the two atropoisomers.14 Crystals of (RP,RP,Sa)-5 were dissolved in deuterated chloroform in a NMR tube, and successive 1H-NMR spectra were collected at specific times.15 After approximately 30 min, the concentrations of the two atropoisomers were the same. The kinetic data followed a first order profile for the Sa/Ra equilibrium with kinetic constant k1 = k − 1 = 1.47 × 10−3 s−1, and a ΔG‡ of 21 kcal mol−1 = 88 kJ mol−1. It is commonly assumed that the free energy barrier between conformational and configurational isomers is 100 kJ mol−1, which means that if the interconversion barrier is higher than 100 kJ mol−1, isomers can be isolated. This finding is in agreement with the observation that atropoisomers (RP,RP,Sa)-5 and (RP,RP,Ra)-5 can be detected by TLC and NMR but cannot be isolated using chromatographic techniques at room temperature.
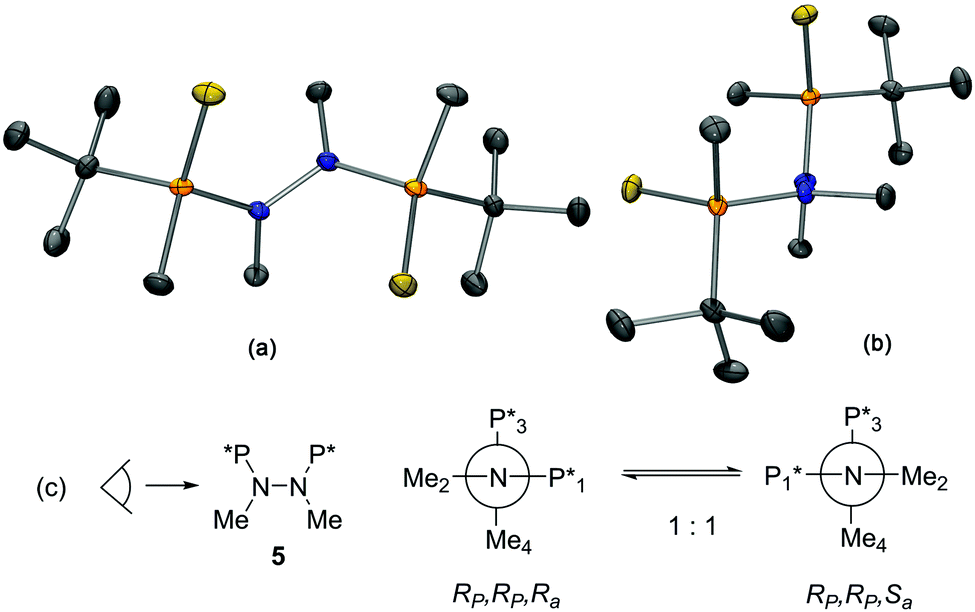 | ||
| Fig. 1 X-Ray structure of (RP,RP,Sa)-5. (a) Perspective and (b) Newman projection through the N–N axis. ORTEP drawing showing thermal ellipsoids at 50% probability. (c) Newman projections for the equilibrium of compound 5. In solution, 5 is present as a mixture of diastereomeric N–N atropoisomers. In the solid state, only the (RP,RP,Sa) atropoisomer is observed. | ||
Similar behavior was observed for pyrazolidine 7. When the crude product was eluted through a SiO2 flash column, two isomers in a 1:1 ratio could be clearly separated and isolated, as confirmed by 1H NMR. However, the samples underwent equilibration within 24 h to the initial 1:1 mixture. 1H NMR analysis showed that, upon crystallization, the crystals contained a single isomer of 7. X-ray diffraction analysis revealed that the unit cell contained two independent molecules of (RP,RN,RN,RP)-7, both bearing the phosphorus atoms with the R configuration (Fig. 2).12 The sum of the angles around the nitrogen centers added up to 348.6/351.8° and 336.0/334.7°. The geometrical constraints imposed by the five-membered ring force the nitrogen atoms to adopt a trigonal pyramidal geometry, with the bulky phosphorus groups lying at opposite sides of the pyrazolidine ring, with a P–N–N–P torsion angle of 122.8/118.6°. In this circumstance, the isomerization equilibrium can more accurately be described as a double nitrogen inversion rather than atropoisomerism (Fig. 2b). The kinetics of the isomerization of 7 were also studied by 1H NMR. Upon dissolution of the pure isomers in CDCl3, almost complete isomerization was observed after 24 h.15 The kinetic constants were calculated to be k1 = k − 1 = 1.04 × 10−5 s−1, with a ΔG‡ of 24 kcal mol−1 (100.4 kJ mol−1). These findings indicate that the energy barrier between (RP,RN,RN,RP)-7 and (RP,SN,SN,RP)-7 was at the border between configurational and conformational isomers, in agreement with the experimental observations.
 | ||
| Fig. 2 (a) X-Ray structure of (RP,RN,RN,RP)-7. ORTEP drawing showing one of the two independent molecules in the unit cell (thermal ellipsoids at 50% probability). (b) Newman projections of the equilibrium for compound 7. In solution, compound 7 is present as a mixture of diastereomers resulting from a double nitrogen inversion. | ||
To date, the use of hydrazine as a backbone for non-chiral and chiral diphosphines and in catalysis has met limited success.16 For instance, chiral hydrazine diphosphoramidites have been described, and their use in the rhodium-catalyzed hydrogenation of Z-MAC afforded up to 93% enantiomeric excess.17 With the aim to study the ability of ligands 5 and 7 in catalysis, we proceeded to test them in the Rh catalyzed asymmetric hydrogenation of standard test substrates.
Heating 5 and 7 with neat pyrrolidine at 80 °C for 2 h resulted in complete borane deprotection.18 After pyrrolidine removal, the crude ligand was reacted with [Rh(COD)2][BArF4] to furnish the desired cationic complexes 8 and 9 (Scheme 4).12,19,20 Asymmetric hydrogenation with catalysts 8 and 9 at 3 bar of H2 and 1 mol% of catalyst loading took place with complete conversions (Table 1). In the reduction of methyl α-acetamidoacrylate (MAA) catalyst 9, with a pyrazolidine backbone, provided better enantioselectivity (98% vs. 95% ee, Table 1, entries 1 and 2). (Z)-Methyl α-acetamido-3-phenylacrylate (Z-MAC) was hydrogenated to full conversion and almost complete selectivity (99% ee) with both catalysts (Table 1, entries 3 and 4). The novel complexes were also active in the hydrogenation of N-(1-phenylvinyl)acetamide (PVA) yielding full conversion and enantiomeric excess up to 97% (Table 1, entries 6 and 7). Reducing the amount of the catalyst to 0.1 mol% of 9 did not have an adverse effect on the selectivity of the reaction (Table 1, entry 8). Finally, catalyst 9 proved to be highly active and selective in the hydrogenation of dimethyl itaconate (DMI), affording the product with full stereocontrol (99% ee, Table 1, entry 9). When the catalyst loading was reduced to 0.02 mol%, a slight decrease in selectivity was observed, and dimethyl itaconate was isolated in 97% enantiomeric excess (Table 1, entry 10).
 | ||
| Scheme 4 Borane removal and complexation to Rh(I). | ||
| Entry | Catalyst (S/C) | Substrateb | Conv.c (%) | eed (%) |
|---|---|---|---|---|
| a Reaction conditions: 0.7 mmol of substrate and 1 mol% of catalyst in dry MeOH were placed in a Büchi 250 Miniclave. The system was charged with 3.0 bar of hydrogen gas and the mixture was stirred for 12 h at r.t. b Methyl α-acetamidoacrylate (MAA), (Z)-methyl α-acetamido-3-phenylacrylate (Z-MAC), (Z)-methyl α-acetamido-3-(3,4,5-trimethylphenyl)acrylate (Z-MATC), N-(1-phenylvinyl)acetamide (PVA), dimethyl itaconate (DMI). c Conversion was determined by 1H NMR of the crude reaction mixture. d Enantiomeric excess determined by chiral GC or HPLC. | ||||
| 1 | 8 (100) | MAA | 99 | 95 |
| 2 | 9 (100) | MAA | 99 | 98 |
| 3 | 8 (100) | Z-MAC | 99 | 99 |
| 4 | 9 (100) | Z-MAC | 99 | 99 |
| 5 | 9 (100) | Z-MATC | 99 | 99 |
| 6 | 8 (100) | PVA | 99 | 95 |
| 7 | 9 (100) | PVA | 99 | 97 |
| 8 | 9 (1000) | PVA | 99 | 97 |
| 9 | 9 (100) | DMI | 99 | 99 |
| 10 | 9 (5000) | DMI | 99 | 97 |
In summary, we report a new class of P-stereogenic C2-symmetric ligands with a hydrazine backbone. The ligands were assembled from hydrazine and optically pure tert-butylmethylphosphinous acid in a rapid and efficient synthesis. The borane-protected ligands 5 and 7 were present as a mixture of atropo- and nitrogen-inversion isomers which are in slow exchange in solution. The nature of such an equilibrium was studied through a combination X-ray crystallography and reaction kinetics. The new ligands were tested in the Rh-catalyzed asymmetric hydrogenation of benchmark substrates demonstrating that ligands with a hydrazine backbone can show excellent catalytic performance.
We thank the MINECO (CTQ2014-56361-P) and IRB Barcelona for financial support. A. P. thanks AGAUR for a Beatriu de Pinós fellowship. IRB Barcelona is the recipient of a Severo Ochoa Award of Excellence from MINECO (Government of Spain).
Notes and references
- (a) Asymmetric Catalysis On Industrial Scale: Challenges, Approaches and Solutions, ed. H. A. Blaser and H. J. Federsel, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed; (b) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS PubMed; (c) L. A. Saudan, Acc. Chem. Res., 2007, 40, 1309 CrossRef CAS PubMed; (d) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS PubMed; (e) A. M. Palmer and A. Zanotti-Gerosa, Curr. Opin. Drug Discovery Dev., 2010, 13, 698 CAS; (f) P. Etayo and A. Vidal-Ferran, Chem. Soc. Rev., 2013, 42, 728 RSC.
- (a) Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (b) W. J. Tang and X. M. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (c) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed; (d) G. Erre, S. Enthaler, K. Junge, S. Gladiali and M. Beller, Coord. Chem. Rev., 2008, 252, 471 CrossRef CAS; (e) H.-U. Blaser, B. Pugin, F. Spindler and M. Thommen, Acc. Chem. Res., 2007, 40, 1240 CrossRef CAS PubMed; (f) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef PubMed; (g) O. I. Kolodiazhnyi, Phosphorus Chemistry I: Asymmetric Synthesis and Bioactive Compounds, in Topics in Current Chemistry, ed. J. Montchamp, 2015, vol. 360, p. 161 Search PubMed.
- H. B. Kagan and T. P. Dang, J. Am. Chem. Soc., 1972, 94, 6429 CrossRef CAS.
- W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567 CrossRef CAS.
- For reviews on P-stereogenic phosphorus compounds, see: (a) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS; (b) O. I. Kolodiazhnyi, Top. Curr. Chem., 2015, 360, 1616 Search PubMed; (c) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771 RSC.
- For selected applications of this family of phosphines and other P-stereogenic phosphines, see: (a) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed; (b) I.-H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS PubMed; (c) H. Ito, T. Okura, K. Matsuura and M. Sawamura, Angew. Chem., Int. Ed., 2010, 49, 560 CrossRef CAS PubMed; (d) X. Wang and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 19080 CrossRef CAS PubMed; (e) Q. Hu, Z. Zhang, Y. Liu, T. Imamoto and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 2260 CrossRef CAS PubMed; (f) Y. Shibata and K. Tanaka, J. Am. Chem. Soc., 2009, 131, 12552 CrossRef CAS PubMed; (g) A. Yanagisawa, S. Takeshita, Y. Izumi and K. Yoshida, J. Am. Chem. Soc., 2010, 132, 5328 CrossRef CAS PubMed; (h) H. Li, K. M. Belyk, J. Yin, Q. Chen, A. Hyde, Y. Ji, S. Oliver, M. T. Tudge, L.-C. Campeau and K. R. Campos, J. Am. Chem. Soc., 2015, 137, 13728 CrossRef CAS PubMed; (i) R. Tan, X. Zheng, B. Qu, C. A. Sader, K. R. Fandrick, C. H. Senanayake and X. Zhang, Org. Lett., 2016, 18, 3346 CrossRef CAS PubMed.
- (a) M. Revés, C. Ferrer, T. León, S. Doran, P. Etayo, A. Vidal-Ferran, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2010, 49, 9452 CrossRef PubMed; (b) T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740 CrossRef PubMed; (c) S. Orgué, A. Flores, M. Biosca, O. Pàmies, M. Diéguez, A. Riera and X. Verdaguer, Chem. Commun., 2015, 51, 17548 RSC.
- E. Cristóbal-Lecina, P. Etayo, S. Doran, M. Revés, P. Martín-Gago, A. Grabulosa, A. R. Costantino, A. Vidal-Ferran, A. Riera and X. Verdaguer, Adv. Synth. Catal., 2014, 356, 795 CrossRef.
- E. Salomó, S. Orgué, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2016, 55, 7988 CrossRef PubMed.
- (a) T. Imamoto, J. Watanabe, Y. Wada, H. Masuda, H. Yamada, H. Tsuruta, S. Matsukawa and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1635 CrossRef CAS; (b) I. D. Gridnev, M. Yasutake, N. Higashi and T. Imamoto, J. Am. Chem. Soc., 2001, 123, 5268 CrossRef CAS PubMed; (c) I. D. Gridnev, Y. Yamanoi, N. Higashi, H. Tsuruta, M. Yasutake and T. Imamoto, Adv. Synth. Catal., 2001, 343, 118 CrossRef CAS.
- A. N. Kornev, N. V Belina, V. V Sushev, G. K. Fukin, E. V Baranov, Y. A. Kurskiy, A. I. Poddelskii, G. A. Abakumov, P. Loennecke and E. Hey-Hawkins, Inorg. Chem., 2009, 48, 5574 CrossRef CAS PubMed.
- CCDC 1533430 (5), 1533431 (7) and 1533432 (9).
- To our knowledge there are almost no examples of N–N atropoisomerism equilibria. For examples of non-biaryl atropoisomers, see: E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239 CrossRef CAS PubMed.
- Since both nitrogen atoms are completely planar, the new source of chirality in 5 is an N–N chiral axis. However, it is possible that in the process of isomerization either one or the two nitrogen atoms adopt a pyramidal structure.
- See the ESI† for full details.
- (a) B. R. Aluri, N. Peulecke, B. H. Müller, S. Peitz, A. Spannenberg, M. Hapke and U. Rosenthal, Organometallics, 2010, 29, 226 CrossRef CAS; (b) A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712 CrossRef CAS PubMed; (c) A. M. Z. Slawin, M. Wainwright and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2002, 513 RSC.
- L. Eberhardt, D. Armspach, D. Matt, B. Oswald and L. Toupet, Org. Biomol. Chem., 2007, 5, 3340 CAS.
- Compound 4 was deboronated in the same conditions. See the ESI† for experimental details and NMR data on the resulting secondary diphosphazene.
- Initial coordination studies were performed using [Rh(COD)2][BF4] as metal precursor but a mixture of the desired product and a cationic rhodium complex bearing two bisphosphine ligands was obtained without possibility to separate them.
- Complex 9 does not show any dynamic behavior and in solution behaves as single C2-symmetric compound. X-ray analysis (ESI†) shows a bite-angle of 82.2° which is slightly smaller than the corresponding carbon analog bisP*-Rh (83.3°), see ref. 10.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and NMR spectra for new compounds; crystallographic data file in CIF format. CCDC 1533430–1533432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc01944k |
| This journal is © The Royal Society of Chemistry 2017 |