
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arene C–H activation by gold(III): solvent-enabled proton shuttling, and observation of a pre-metallation Au–arene intermediate†
L.
Rocchigiani
a,
J.
Fernandez-Cestau
a,
P. H. M.
Budzelaar
*b and
M.
Bochmann
 *a
*a
aSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: m.bochmann@uea.ac.uk
bDepartment of Chemical Sciences, Federico II University of Naples, Via Cintia, 80126 Napoli, Italy. E-mail: p.budzelaar@unina.it
First published on 3rd April 2017
Abstract
Selective Au–C bond cleavage and arene–C–H activation in (C^N^C)Au(III) pincer complexes are reversible, leading to a solvent-dependent proton shuttling process. The ether-free cleavage products are non-fluxional and show weak gold(III)–arene interactions commensurate with intermediates postulated for CMD-type arene activation.
The metalation of arene C–H bonds is an important synthetic tool. Since these reactions are frequently highly site-selective, they have become a method of choice for C–C bond formations and arene functionalizations.1 Compared to other noble metals, the C–H activation by gold and formation of cyclometallated gold aryl complexes is synthetically more limited and often very sensitive to the nature of arene substituents. This notwithstanding, cyclometallations form the basis of several types of gold pincer complexes2,3 which find widespread use ranging from photoluminescent compounds4,5 to anti-cancer agents.6
The activation of aromatic C–H bonds by gold trichloride to give arylgold halide complexes was first reported in the 1930s.7 The reaction most probably proceeds by an SEAr pathway and involves attack of an electrophilic metal ion on the arene π-system, followed by HCl elimination. This mechanism is characterized by a zwitterionic Wheland intermediate in which there is significant distortion of the C–H bond angle (Scheme 1a). Since it proceeds via a cationic intermediate, this path is favoured by electron donating ring substituents.8 On the other hand, basic gold complexes react smoothly with fluorinated arenes to give the corresponding gold aryl products.9,10 For reactions of this type, an alternative mechanism is favoured: Concerted Metallation-Deprotonation (CMD). This pathway is well established for arylations and cyclometallations of palladium,11 and is characterized by only small angular distortion of the arene C–H bond and weak π-interaction between the metal and the arene ring (Scheme 1b). The metal–arene interaction needs to be just sufficiently strong to increase the acidity of the C–H bond to facilitate abstraction by a base, such as acetate. CMD reactions have been probed mechanistically and computationally and, in the case of metal acetates, involve a 6-membered cyclic intermediate. For palladium this mechanism has been demonstrated for a wide range of arenes and heteroarenes.12,13
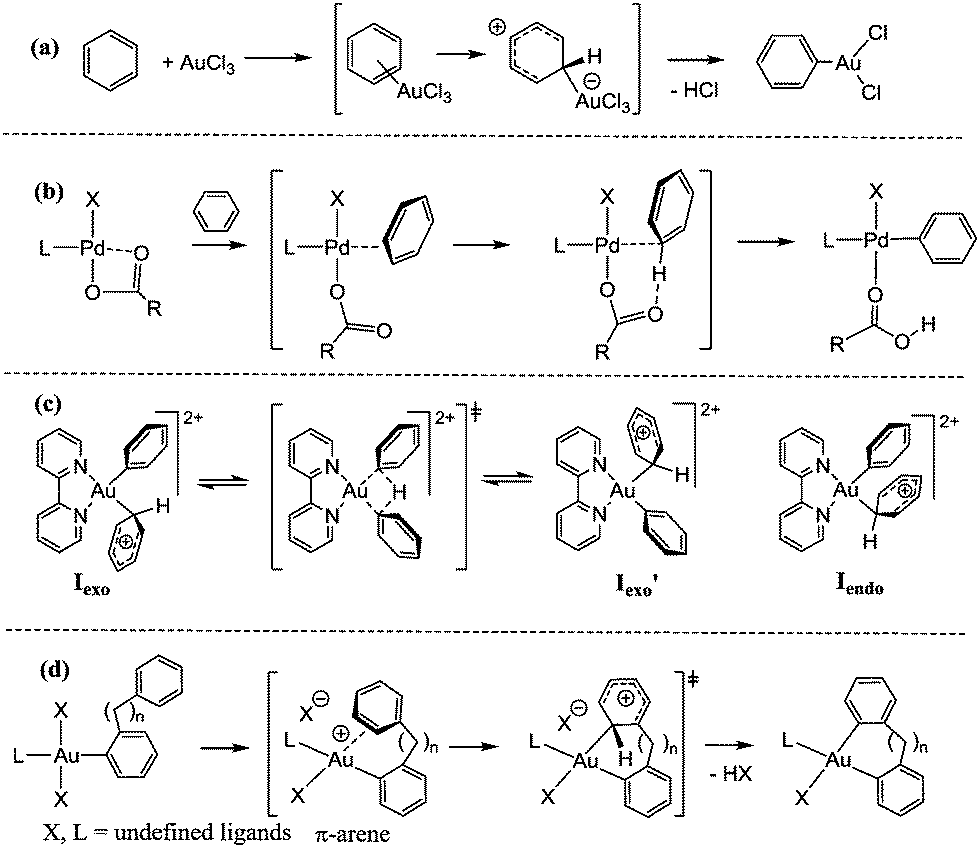 | ||
| Scheme 1 (a) Arene auration by AuCl3via the SEAr mechanism;7 (b) arene metalation by concerted metalation–deprotonation (CMD);11–13 (c) hypothetical degenerate H-exchange in an AuIII(phenyl)(benzene) model complex;14 (d) proposed arene C–H activation pathway in AuIII-catalysed arene–arylsilane coupling.15 | ||
To the best of our knowledge, for gold(III) there is only one report of a computational investigation of the arene metallation mechanism, on the solvent-free reversible activation of benzene by the [(bipy)AuPh]2+ dication. In this case an energy minimum for an η2-benzene adduct could not be located, but Wheland-type η1 intermediates are thought to be formed (Scheme 1c). A proton exchange pathway involving the two ipso-carbon atoms was found to be energetically slightly favoured, with a reaction barrier of 7.8 kcal mol−1 with reference to the exo intermediate Iexo. However, no experimental evidence for benzene activation by [(bipy)AuPh(L)]2+ (L = weakly coordinating ligand) or [(bipy)AuPh2]+ could be found.14 Similar processes have been invoked in catalytic and kinetic studies on gold(III) mediated intra- and intermolecular oxidative aryl–aryl coupling reactions of arylsilanes. The intramolecular version of this reaction proved particularly amenable to detailed kinetic investigations, and on the basis of Hammett relationships, an SEAr-type arene activation mechanism was preferred (Scheme 1d).15
Instructive as such kinetic studies are, the nature of intermediates and transitions states must remain a matter of conjecture, and the ligand sphere of the gold species thought to be involved in catalysis remains ill-defined. We set out therefore to study the intramolecular activation of arenes in a well-defined system, based on the selective Au–C bond cleavage of (C^N^C)AuX pincer complexes10 with acid (C^N^C = 2,6-(C6H3But)2pyridine dianion). We showed earlier that such pincer complexes react with trifluoroacetic acid under selective cleavage of one of the Au–C bonds to give the chelate (C^N-CH)Au(X)(OAcF).16 We report here the Au–C cleavage reaction of (C^N^C)AuX (1a, X = Cl; 1b, X = C6F5) with the Brønsted acid [H(OEt2)2]+[H2N{B(C6F5)3}2]− (HAB2),17 to create highly reactive [(C^N-CH)AuX]+ cations. Here the anion does not compete for metal coordination sites, so that weak metal–ligand interactions can be observed.
The reactions of 1a, b with 1 equiv HAB2 in CD2Cl2 afford the protodeauration products [(C^N-CH)AuX(OEt2)]+AB2− (2a, b) in quantitative yields. Full NMR characterization supported the formulation as ion pairs with non-coordinating amido-diborate counterions. Diffusion NMR experiments demonstrated the monomeric nature of the cations and Et2O coordination; the latter was further confirmed by the presence of selective dipolar interactions between Et2O protons and aromatic resonances in the 1H NOESY NMR spectrum (see ESI,† Fig. S11–S13).
Rather unexpectedly, analysis of the dipolar contacts in the NOE spectra of 2a and 2b revealed a selective chemical exchange process between protons 5′ of the protodeaurated aryl ring with protons 5 of the metal-bound phenyl (Scheme 2 and ESI,† Fig. S5 and S6). This observation suggests that protodeauration is reversible on the NOE timescale, due to the efficient C–H activation of the pendent aryl ring, which regenerates a transient concentration of 1, together with the release of the acid.
 | ||
| Scheme 2 Ether-mediated reversible Au–C protonation/C–H deprotonation, showing the atomic numbering used for NMR assignments. | ||
For 2a at 323 K the rate of exchange kEX(2a) was 2.3 s−1, leading to an exchange barrier of ΔG‡ = 18.3 kcal mol−1 (1H EXSY NMR, C6D5Cl). An Eyring analysis over T = 298–343 K allowed the determination of ΔH‡ = 11 kcal mol−1 and a negative activation entropy, ΔS‡ = −23 cal mol−1 K−1. The rate of exchange of the perfluorophenyl complex 2b was slightly slower, kEX(2b) = 1.1 s−1 (ΔG‡ = 18.8 kcal mol−1 at 323 K). Other cationic Au–aryl complexes such as 1c (X = p-C6H4F) or 1d (X = C6H5) gave analogous cleavage products 2c, 2d but without any indication of reversibility on the NOE timescale, which suggests that in these cases cyclometallation is more difficult (ΔΔG‡ ≥ 3.5 kcal mol−1, considering an NOE-observation limit of kEX ≈ 0.01 s−1).
Generating 2b in chlorobenzene-d5 at room temperature followed by several vacuum/CD2Cl2 cycles gives solutions which are essentially free of diethyl ether. The resulting changes in the NMR spectra, especially of the signals of the “dangling” C6H4But substituent, are consistent with the conversion of 2b to a new species 3b. Thus the unremarkable 13C NMR chemical shifts of the o- and m-C atoms in 2b (C5′, C6′, respectively, Scheme 3) move from δC = 127.0 and 129.3 to δC = 119.3 (C5′) and 138.2 ppm (C6′) in 3b. On the other hand, there is no major change in 1JC,H values (2b: 1JC,H = 160 Hz for C5′ and C6′; 3b: 1JC,H = 159 (C5′) and 167 Hz (C6′)).
 | ||
| Scheme 3 Generation of ether-free gold cations indicating changes in the 13C NMR data of the aryl substituent (1JC,H values in italics). | ||
The data are consistent with an interaction between the gold ion and the “dangling” aryl side-arm. The C^N-CH ligand system is too strained to allow η2-type π-coordination of C6H4But to the metal centre. Steric factors also dictate that the only way the C6H4But moiety in 3b can approach the metal is by tilting by about 45° relative to the (C^N)Au plane, to maximize the interaction of Au with one of the ortho-C atoms (C5′). Since the bilateral symmetry of C6H4But is retained in the 1H NMR spectrum, there must be rapid interchange between the two ortho positions, which suggest that the Au···arene interaction is rather weak. Consistent with this, VT 1H NMR spectra down to −85 °C showed broadening of the H5′ resonance but no loss of symmetry. Evidently the tilting motion is not frozen at that temperature, so that gold alternately interacts with both C5′ atoms.‡ Thus the metal polarizes the aryl substituent, but without significant deformation of the aryl C–H bond which would result in changes in the 1JC,H coupling constant. Such an interaction is most in line with the model proposed for a CMD intermediate (Scheme 1b). It appears therefore that with 3b it has been possible to generate such a species as a local minimum structure.
Another instructive difference between the ether complex 2b and ether-free 3b concerns the proton shuttling process. Whereas in 2b proton exchange leads to reversible C–H activation, as described above, ether-free complexes 3a, 3b show no H+ mobility: protodeauration is no longer reversible. As has been amply demonstrated kinetically and in computational models, in palladium-mediated C–H activations by the CMD mechanism it is the bridging acetate that enables the deprotonation step.11–13 The facile proton exchange in gold complexes of type 2 shows that this role can also be fulfilled by a weakly basic solvent: at least one ether molecule is required for proton shuttling. In its absence, Au–C cleavage becomes irreversible. To our knowledge this is the first clear illustration of the role of even weakly binding solvents in C–H activation processes in gold chemistry.
The observed proton shuttling, the role of solvent, and the proposed structure of a CMD-type arene complex were further supported by modelling studies using density functional theory (DFT) calculations. The mechanism of proton exchange in 2a, the reaction pathway and its energy profile are shown in Scheme 4.
 | ||
| Scheme 4 Reversible deprotonation pathway for (C^N^C)AuCl with calculated stationary points for TS-D and species E (top) and calculated free-energy profile [bold: ΔG (kcal mol−1); in italics: ΔH (kcal mol−1); ΔS (cal mol−1 K−1)]. | ||
At the starting point, the oxidative protonation of an AuIII complex to a cationic AuV intermediate was checked but quickly ruled out. We therefore focused on direct Au–C protonation. In order to explore mechanistic variations, we used a simplified model ligand lacking the tBu substituents and H(OMe2)2+ as a model for [H(OEt2)2]+[H2N{B(C6F5)3}2]−, to minimise conformational issues. Structures were fully optimized in the presence of a solvent model (chlorobenzene, PCM model of Gaussian09).
It was assumed that the acid H(OR2)2+ approaches the neutral complex A to give the association complex Cvia transition state TS-B. The formation of TS-B is associated with the negative entropy that was experimentally observed for the overall process, since the path from C to F is essentially entropy-neutral. In C the acidic O–H bond has already approached the Au–C bond quite closely (H–O = 1.01 Å; H···C = 1.93 Å). From there, the proton is transferred to carbon with a very small barrier (≤1 kcal mol−1, viaTS-D) to produce three-coordinate complex E, releasing the conjugate base OR2. This intermediate may then dimerize to G, although the energetically preferred pathway is to capture OR2 to form the ether complex F.
Further checks showed that the use of OEt2 instead of OMe2 as a base reduces the exchange barrier by 3–4 kcal mol−1. About half of that (∼2 kcal mol−1) is due to the lower complexation energy (presumably for steric reasons) of OEt2 to Au; the remainder is likely related to the higher Brønsted basicity of OEt2 compared to OMe2.
There is an equivalent pathway which assumes dissociation of H(OR2)2+ into HOR2+ + OR2 prior to attack on complex A. However, this would not explain the observed negative activation entropy.
The potential-energy surface is rather flat around the relevant species C + OR2, TS-B and A + H(OR2)2+, but a stationary point with the correct geometry and reaction coordinate for such a proton transfer could be located. In this scenario TS-B is (barely) rate-limiting. The calculated effective activation parameters for the simplified model system (ΔH‡ = 11.5 kcal mol−1, ΔS‡ = −24.9 cal mol−1 K−1, ΔG‡ = 19.5 kcal mol−1 at 50 °C) agree well with the ones derived from the Eyring analysis for the real system. Over the stages TS-D⇌C⇌TS-B the free-energy profile is virtually flat (within 2 kcal mol−1), but the partitioning in ΔH‡ and TΔS‡ varies considerably.
Alternatively, the acid complex C could rearrange to have the O–H bond interact with the second Au-bound carbon (a TS could not be located), which would lead to a presumably somewhat lower barrier of at least 19 kcal mol−1 for the intramolecular proton exchange, in close agreement with the observation. On the other hand, intermolecular proton exchange would involve formation of A and free HOR2+ as the highest-energy point of the reaction, with a calculated ΔG(F–A + HOR2+) of ∼25 kcal mol−1, which is somewhat higher than was observed experimentally.
The effect of substituents X on the proton exchange reaction was explored assuming that the path involving TS-B shown in Scheme 4 can be extended to variations of the Au complex, and that in all cases the variation in free energy over these structures is small. This allows us to use the free energy difference ΔG‡ (F–TS-D) as a first estimate of the effective deprotonation barrier (see ESI,† Table S4 for a summary of the predicted substituent effects on deprotonation barriers).
For complexes bearing aryl groups at Au one could also envisage competing protonation of the non-chelated Au–aryl bond. The barrier calculated for this process (for X = C6H5) is ∼7 kcal mol−1 higher than for protonation at the C^N^C ligand, in agreement with the observed selective protonation of the Au–C bond of the pincer ligand. Two factors that are important here are the ring strain in the (C^N^C)Au framework (promoting ligand protonation) and the out-of-plane orientation of the aryl group at Au (hindering its protonation).
The possible structure of the ether-free complex 3b was also interrogated by DFT methods. No evidence was found for agostic Au···H–C bonding18 or a π-complex. The optimized structure converges to a situation with rather long contacts with the metal, Au-ortho-C 2.81 Å and Au–H 2.61 Å and no evidence for a strong Au–(CH) interaction; the H atom is bent out of plane of the 6-ring by less than 10 degrees. Calculations of the 13C NMR chemical shifts for ether-bound 2b and solvent-free 3b confirmed the observed chemical shift trends (with the simplification that OMe2 was used in the model). For 2b, 13C shifts in the range expected for a non-bound phenyl group were calculated, δ 129.47 and 128.23 for the C5′ and C6′ signals of –C6H4But, respectively. In the model of solvent-free 3b, with the –C6H4But substituent tilted by 45° relative to the plane through the (C^N)Au chelate, shifts of δ 117.5 and 130.7 (C5, C5′; average 124.1) and 133.29 and 135.76 (C6, C6′; average 134.5) were determined, which qualitatively reflect the observed increase in Δδ between the o- and m-C chemical shifts. On rotation of the ring to 90° these differences disappear. According to Wiberg bond indices, in the naked [(C^N-CH)Au]+ ion the interaction of Au with the nearby ortho-carbon atom amounts to about 10% of a full covalent Au–C bond (using the true Au–C bond as reference). This interaction reduces to about 1–2% on rotation of the phenyl ring to a perpendicular orientation, or on coordination of OMe2 to the empty site. The direct interaction of Au with the ortho-hydrogen is always very small. The calculations appear to slightly underestimate the metal–arene interaction compared to the experimental NMR data but are generally consistent with observation. The best model appears to be the type of incipient near-perpendicular metal–arene interactions envisaged in the CMD mechanism (Scheme 1b), whereas the data do not support the formation of a Wheland-type intermediate. In the active proton shuttling process such structures would be close to the transition state. In the absence of a solvent to provide H+ mobility, weak metal–arene bonding becomes a resting state.
In summary, we show here that (i) both Au–C bond cleavage and C–H activation in well-defined gold(III) systems can be both facile and reversible; (ii) proton shuttling depends on the presence of a weakly basic solvent, in this case ether, which facilitates proton abstraction from a metal-bound arene; (iii) in the absence of such a solvent, proton exchange is arrested and a structure with a polarized aryl through gold–arene π-interaction becomes the spectroscopically detectable ground state. Metal–arene interactions of this type have previously been proposed for C–H activation processes by the Concerted Metallation-Deprotonation (CMD) mechanism. To our knowledge, the present results provide the first experimental evidence for such interactions.
This work was supported by the European Research Council. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT).
Notes and references
- (a) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef CAS PubMed; (b) M. Albrecht, Chem. Rev., 2010, 110, 576 CrossRef CAS PubMed; (c) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (e) Z. X. Huang, H. N. Lim, F. Y. Mo, M. C. Young and G. B. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (f) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed.
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 2 CrossRef.
- D.-A. Roşca, J. A. Wright and M. Bochmann, Dalton Trans., 2015, 44, 20785 RSC.
- (a) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed; (b) G. S. M. Tong, K. T. Chan, X. Y. Chang and C.-M. Che, Chem. Sci., 2015, 6, 3026 RSC; (c) A. Szentkuti, M. Bachmann, J. A. Garg, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2014, 20, 2585 CrossRef CAS PubMed.
- (a) J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629 RSC; (b) L. Currie, J. Fernandez-Cestau, L. Rocchigiani, B. Bertrand, S. J. Lancaster, D. L. Hughes, H. Duckworth, S. T. E. Jones, D. Credgington, T. J. Penfold and M. Bochmann, Chem. – Eur. J., 2017, 23, 105 CrossRef CAS PubMed.
- (a) B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209 RSC; (b) T. T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786 RSC.
- (a) M. S. Kharasch and H. S. Isbell, J. Am. Chem. Soc., 1931, 53, 3053 CrossRef CAS; (b) Y. Fuchita, Y. Utsunomiys and M. Yasutake, J. Chem. Soc., Dalton Trans., 2001, 2330 RSC.
- Y. Liu, Z. Z. Yu, J. Z. H. Zhang, L. Liu, F. Xia and J. L. Zhang, Chem. Sci., 2016, 7, 1988 RSC.
- (a) P. F. Lu, T. C. Boorman, A. M. Z. Slawin and I. Larrosa, J. Am. Chem. Soc., 2010, 132, 5580 CrossRef CAS PubMed; (b) I. I. F. Boogaerts and S. P. Nolan, Chem. Commun., 2011, 47, 3021 RSC , and cited references.
- D.-A. Roşca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247 RSC.
- D. Garcia-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658 CrossRef CAS PubMed.
- M. K. Ghosh, M. Tilset, A. Venugopal, R. H. Heyn and O. Swang, J. Phys. Chem. A, 2010, 114, 8135 CrossRef CAS PubMed.
- (a) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (b) T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245 CrossRef CAS PubMed.
- D. A. Smith, D.-A. Roşca and M. Bochmann, Organometallics, 2012, 31, 5998 CrossRef CAS.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. L. Hughes and M. Bochmann, Organometallics, 2002, 21, 451 CrossRef CAS.
- H. Schmidbaur, H. G. Raubenheimer and L. Dobrzańska, Chem. Soc. Rev., 2014, 43, 345 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR spectroscopic and computational details. See DOI: 10.1039/c7cc01628j |
| ‡ The aryl oscillation implies that the observed NMR parameters of C5′ and C6′ in 3b are average values. |
| This journal is © The Royal Society of Chemistry 2017 |