
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Initiating radical reactions with non-thermal plasmas†
Yury
Gorbanev
ab,
Dirk
Leifert
c,
Armido
Studer
 c,
Deborah
O'Connell
*b and
Victor
Chechik
*a
c,
Deborah
O'Connell
*b and
Victor
Chechik
*a
aDepartment of Chemistry, University of York, York YO10 5DD, UK. E-mail: victor.chechik@york.ac.uk
bYork Plasma Institute, Department of Physics, University of York, York YO10 5DD, UK
cOrganic Chemistry Institute, University of Münster, Corrensstrasse 40, D-48149, Münster, Germany
First published on 6th March 2017
Abstract
Non-thermal plasmas were used as radical initiators in three radical chain reactions: dehalogenation, 5-exo-trig cyclisation and trifluoromethylation with the Togni II reagent. The use of non-volatile solvents (or solvent-free conditions) gave high yields in all reactions under optimised conditions. Short reaction times and mild conditions make this method an attractive alternative to conventional radical initiators.
Recently developed non-thermal plasmas, operating at atmospheric pressure and near room temperature, can be used as sources of reactive species generated under very mild conditions.1,2 However, typical concentrations of free radicals in non-thermal plasmas (ca. 1010–1014 molecules cm−3)3 are usually too low for stoichiometric radical reactions on a reasonable scale.4 For instance, a stoichiometric radical reaction on a 0.01 mole scale would require exposure to 105–109 L of plasma gas, which is impractical for synthetic applications. The situation is different for the initiation of radical chain reactions (e.g., polymerisation), where only a small amount of an initiator is required to overcome termination processes. For these reactions, non-thermal plasmas can be a viable alternative to other types of initiation.2 Plasma initiation is attractive due to mild reaction conditions and the absence of exogenous initiators (often toxic or hazardous, e.g., peroxides, azo derivatives, transition metal salts, etc.). The low level of UV radiation generated in these plasmas does not produce a detectable amount of free radicals,5,6 so plasma initiation can be used with photosensitive substrates. The low concentration of plasma-derived radicals limits damage to the substrates and products, and the overall process is controlled by the substrate/reagent reactivity rather than the properties of the initiator radicals.
Here, we explore the feasibility of using non-thermal plasmas for synthetic radical chemistry. The reactions are initiated by free radicals (e.g., H˙, ˙OH, O2˙−), formed in the plasma from water/oxygen additives or impurities in the feed gas and diffused into the liquid phase.6 Three reactions were chosen in this study to cover a range of solvents and reaction conditions, different types of chain transfer agents, and intra- and intermolecular pathways. Two plasma sources were used: (a) a cross-field nanosecond (NS) pulsed plasma operated at 40 kV, 3.5 kHz, with N2, He or air as a feed gas, with the reaction mixture introduced directly into the plasma, and (b) a parallel-field kHz sinusoidal plasma operated with He at ca. 18 kV, 25 kHz, with the reaction mixture exposed to the plasma effluent (see the ESI† for further details). The NS plasma (short pulses repeated with a frequency of 3.5 kHz) is less prone to heating. In our plasma set-ups, both solvated electrons and free radicals could contribute to the initiation.
A general plasma set-up is shown in Fig. 1. The initiating species are formed in the gas phase; efficient mass transfer is thus key to successful initiation. To achieve good mixing, the experiments included recirculation of the reaction mixture through the plasma chamber, a solvent-free reaction and a reaction involving mechanical agitation (stirring).
 | ||
| Fig. 1 A general representation of the plasma setup. The optional components are shown with dashed lines. | ||
In the first case study, we explored dehalogenation of iodo-substituted benzoates7 with sodium dibasic hypophosphite as a non-volatile H donor/chain transfer agent (Scheme 1). Due to exposure to a significant gas flow (typically 1–10 L min−1), plasma initiated reactions cannot be carried out with volatile solvents, reagents or products. In this example, an aqueous system was used. The likely initiators were H˙ and ˙OH radicals generated by plasma in water.
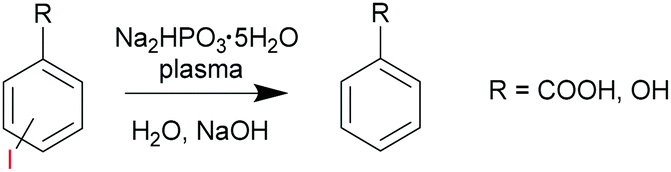 | ||
| Scheme 1 Dehalogenation of iodo-substituted aromatic compounds via a reaction with sodium dibasic hypophosphite initiated by non-thermal plasma. | ||
Initial experiments used an NS plasma with N2 feed gas. The aqueous reaction mixture was nebulised and the resulting aerosol was passed with the feed gas through the discharge region (Experimental and Fig. S1a in the ESI†). In this system, only a low product yield was observed (ca. 7%, see the ESI†). Similar results were obtained by flowing the reaction mixture down the walls of the plasma chamber. The low yields can probably be attributed to the short residence time in the plasma chamber and insufficient efficiency of the radical chain transfer.
The yields increased significantly when the reaction mixture was continuously re-circulated through the plasma chamber with a peristaltic pump (ESI,† Table S1). The yields increased over time but were independent of the flow rate. Operating the plasma with air instead of nitrogen resulted in no product formation (Fig. 2), as the rapid reaction of intermediate radicals with oxygen breaks the radical chain. The use of He as a feed gas significantly increased the product yield to 52% after 20 min of reaction, while the same reaction under N2 gave a 26% yield. This is likely due to the higher electron density in the helium plasma. However, use of a high flow of helium with long reaction times is impractical.
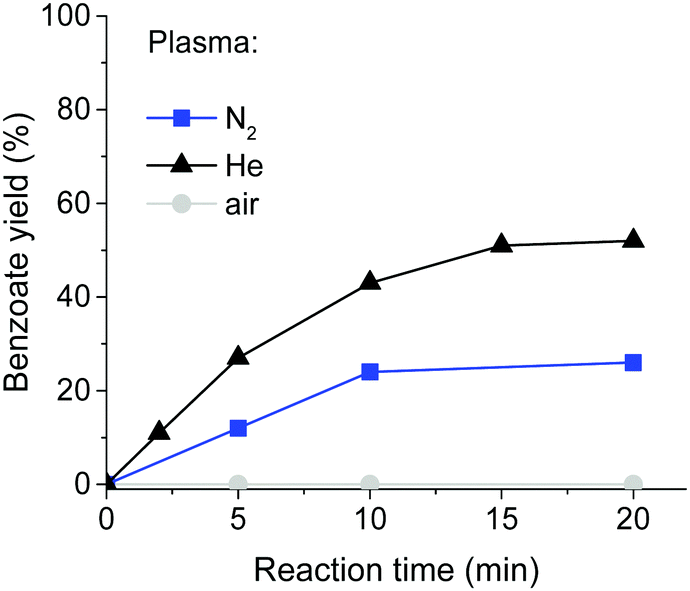 | ||
| Fig. 2 Yield of sodium benzoate in the reactions of 4-iodobenzoate with different plasma gases as determined by 1H-NMR. Reaction conditions: 12 mL of aqueous solution containing 0.6 mmol of 4-iodobenzoic acid, 1 mmol of Na2HPO3·5H2O, and 1.6 mmol of NaOH was exposed to plasma. Plasma parameters: NS, 40 kV, 3.5 kHz, gas flow 10 L min−1. Liquid flow 4 mL min−1. | ||
Interestingly, the reaction stopped after ca. 15 min as no further conversion was observed at longer exposures. At the same time, we observed evaporation of ca. 50% of solvent (water), due to the temperature increase in the continuously sustained plasma. Continuous addition of water to the reaction mixture during the exposure to N2 plasma led to an increased yield of the dehalogenated product over an extended reaction time (see the ESI,† Fig. S2). A similar effect was observed when the concentration of the reagents in the initial reaction mixture was reduced. Under optimised conditions, the product yield (by 1H-NMR of the crude mixture) was ca. 95%, with 79% isolated yield after re-crystallisation.
Other iodo-substituted substrates (2-iodobenzoate, 4-iodophenolate) also afforded good yields in a continuous plasma initiated reaction, whereas dehalogenation was not successful with 4-chlorobenzoate (Fig. S3, ESI†). Exposure of the aqueous solution of 4-iodobenzoate to the plasma with no added hypophosphite afforded a much lower yield of the dehalogenated product (Fig. S4, ESI†). This confirmed that successful plasma initiation is possible only for efficient chain reactions.
The dramatic loss of conversion observed upon solvent evaporation could be due to the increased viscosity, which reduced the diffusion of reactive species from the gas phase into the liquid. In order to test this hypothesis, we added NaCl, Na2SO4 and PEG-300 to the reaction mixture in amounts which corresponded to the viscosity created by sodium iodide formed in the reaction (see Experimental in the ESI†). While NaCl and PEG-300 hindered the reaction, the addition of Na2SO4 did not affect the reaction rate or the yields, despite a similar increase in viscosity (Fig. 3). We thus conclude that the reduced reactivity is not due to the higher viscosity or ionic strength of the solution, but can most probably be attributed to the scavenging of free radicals by iodide ions in concentrated solutions,8 thus breaking the radical chain.
 | ||
| Fig. 3 The effect of viscosity modifiers on the N2 plasma-initiated reduction of 4-iodobenzoate as determined by 1H-NMR. Reaction conditions: 12 mL of aqueous solution containing 0.6 mmol of 4-iodobenzoic acid, 1 mmol of Na2HPO3·5H2O, 1.6 mmol of NaOH, and a viscosity modifier was exposed to plasma. Plasma parameters: NS, 40 kV, 3.5 kHz, gas flow 10 L min−1. Liquid flow 4 mL min−1. | ||
In order to probe the feasibility of using plasma initiation for radical cascade reactions, we studied a 5-exo-trig cyclisation of a vinyloxyethyl radical (Scheme 2). The reaction was carried out in water with added surfactant cetyltrimethylammonium bromide (CTAB) and 1-ethylpiperidine hypophosphite (EPHP) as an H donor/chain transfer reagent.9 When exposed to an NS plasma (N2 or He) with a re-circulation system described above, the yield of the cyclic product did not exceed 5%, with the rest of the starting material remaining unconverted (ESI,† Table S2).
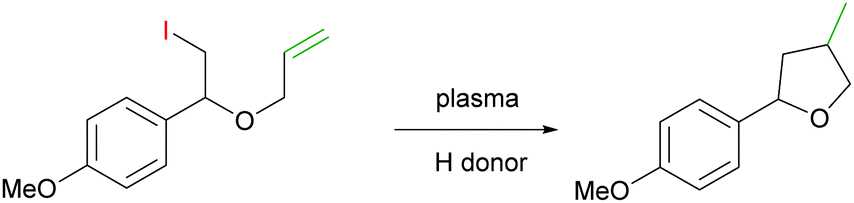 | ||
| Scheme 2 C–C 5-exo-trig radical cyclisation of 2-iodo-1-(4-methoxyphenyl)-1-vinyloxyethane initiated by non-thermal plasma. | ||
To completely exclude oxygen, we carried out the reaction using a static liquid sample exposed to a parallel-field helium-operated plasma in an air-free reactor,10 where the reaction mixture was not circulated through a plasma chamber (Fig. 1; see also Experimental and Fig. S1b in the ESI†). However, the exposure of the H2O/CTAB mixture to the plasma afforded virtually no product (Table S2, ESI†). We tentatively attribute this to the formation of a surfactant film on top of the aqueous solution which prevented efficient diffusion of free radicals from the gas phase into the liquid.11 The other likely reason was a short exposure time (2 min, limited due to solvent evaporation). In general, this type of plasma is prone to heating upon prolonged use, and low boiling solvents including water evaporate rapidly.‡ For instance, deiodination of 4-iodobenzoate under these conditions resulted in almost complete evaporation of the solvent after only 2 min of plasma exposure, affording a 4% product yield (Table S3, ESI†).
To prevent solvent evaporation, we investigated the feasibility of running plasma-initiated reactions solvent-free. The cyclisation reaction (Scheme 2) gave a ca. 5% product yield after 15 min plasma exposure (Table 1, entry 1). Different solvents and H donors/chain transfer agents were screened (Table 1). The reaction was probably initiated by free radicals generated from the solvent and/or impurities, or solvated electrons.1,12,13 The results indicated that when the reaction mixture was not completely homogeneous (i.e., solvent-free, with H donors only partially soluble in the substrate; entries 1 and 8) the yields were low. A similar effect was observed with solvents that did not fully dissolve the H donors (entries 2 and 3). This suggested that the efficient mass transfer is critical for these reactions.
| Entry | Radical transfer agent | Reaction time (min) | Solvent | Yielda (%) | cis/trans |
|---|---|---|---|---|---|
| a Estimated by 1H-NMR analysis. b 82% isolated yield after column purification. c Reaction in air. d Without plasma exposure. | |||||
| 1 | EPHP, 10 eq. | 15 | — | 5 | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :73 :73 |
| 2 | EPHP, 10 eq. | 15 | C6H3(CH3)3 | 2 | 31:69 |
| 3 | EPHP, 10 eq. | 15 | PEG-300 | 4 | 33:67 |
| 4 | (TMS)3SiH, 10 eq. | 15 | — | 88 | 52:48 |
| 5 | (TMS)3SiH, 5 eq. | 20 | — | 96b | 49:51 |
| 6c | (TMS)3SiH, 5 eq. | 20 | — | 32 | 47:53 |
| 7d | (TMS)3SiH, 5 eq. | 180 | — | 0 | — |
| 8 | Ph3SiH, 2 eq. | 15 | — | <1 | — |
An H donor/chain transfer agent tris(trimethylsilyl)silane ((TMS)3SiH) was fully miscible with the substrate. Under optimised conditions (Table 1, entry 5), ca. 96% yield of the cyclic product was achieved (the lower 74% isolated yield is due to the difficult separation of the product from the excess transfer reagent9). A control experiment in which the reaction mixture was allowed to stand inside the He-filled reactor for 3 hours without plasma exposure showed no product formation (entry 7).
In the third example of a plasma-initiated radical chain reaction, we explored radical trifluoromethylation of isonitriles with the Togni-II reagent14 (Scheme 3). Isonitriles are good radical acceptors15 and their radical trifluoromethylation is well established.13,16 Plasma-derived solvated electrons, or free radicals formed from solvent or impurities, could act as initiators for this reaction.
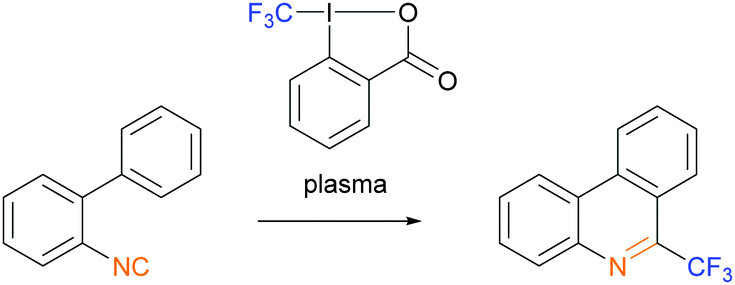 | ||
| Scheme 3 Trifluoromethylation of 2-isocyano-1,1′-biphenyl with the Togni-II reagent as the source of ˙CF3 radicals initiated by plasma. | ||
The initial solvent screening indicated that high boiling point solvents performed best. The use of low molecular weight poly-(ethylene glycol) PEG-300 or tetraethylene glycol (TEG) as solvents afforded the highest yields (Table 2; see also Table S4, ESI†). Solvent-free conditions (Table 2, entry 2) yielded only 2% of the trifluorinated product, while PEG-300 under the same conditions afforded a 17% yield (entry 1). The yields were similar for an air-free reactor and a reaction in an ambient atmosphere (entries 1 and 3), presumably because diffusion of oxygen17 in a He plasma was insufficient for efficient chain termination in this case.18 The ambient atmosphere setup allowed for larger volumes of the reaction mixture to be employed, and the magnetic stirring of the reaction mixture (Fig. 1; see also Fig. S1c, ESI†). When a larger amount of the solvent was used, the yield after 10 min increased from 11 to 25%. Further increase in the solvent volume resulted in a lower yield (entries 3–5). The increased yield in the more dilute solution can be attributed to the limited solubility of the Togni-II reagent in PEG-300 and thus less efficient mass transfer in the more concentrated reaction mixture. The absence of stirring resulted in a lower yield (entry 7), supporting this hypothesis. A 95% crude yield of the 6-(trifluoromethyl)phenanthridine (80% isolated yield) was achieved under optimised conditions.
| Entry | Reaction time (min) | Solvent volume (μL) | Yielda (%) |
|---|---|---|---|
| a Estimated from 19F-NMR spectra. b In an air-free reactor. c With a dielectric foam between the hotplate and the reaction mixture container. d Without stirring. e 1.2 eq. of the Togni-II reagent. f 80% isolated yield. g At 70 °C without plasma exposure. | |||
| 1b | 15 | 200 | 17 |
| 2b | 15 | — | 2 |
| 3 | 10 | 200 | 11 |
| 4 | 10 | 500 | 25 |
| 5 | 10 | 2500 | 20 |
| 6c | 10 | 500 | 14 |
| 7d | 10 | 500 | 17 |
| 8e | 30 | 500 | 95f |
| 9g | 30 | 500 | <1 |
When a glass container with the reaction mixture was positioned directly on top of a metal surface (e.g., a magnetic stirrer plate), the discharge parameters were different from those observed with a glass-only system. While the voltage and frequency of the discharge were kept the same in both cases, it is possible that both the metal surface under the glass container and the ground electrode on the plasma tube served as earth electrodes (Fig. S1c, ESI†). With a dielectric foam layer between the glass container and the metal surface, the yields of the reaction product decreased while the effect of stirring (i.e., enhanced mass transfer) became more pronounced (Table 2, entry 6; and Table S4, ESI†). This suggests that without a dielectric layer, the discharge may have propagated into the liquid.19 Moreover, without the dielectric layer, the temperature of the solution increased to ca. 70 °C after 1 min of plasma exposure (Table S5, ESI†). The system with a dielectric foam layer resulted in a lower temperature after plasma exposure (ca. 60 °C). To check the possibility of a thermally activated reaction, the substrate and the Togni-II reagent were mixed with 500 μL of PEG-300 and kept at 70 °C without plasma exposure. Only a negligible amount of the product was observed (Table 1, entry 9), suggesting the absence of thermal initiation under these conditions. We hypothesise that the increased yield observed for the system without the dielectric foam layer can be attributed to the reduced viscosity of the solvent (PEG-300) at elevated reaction temperature. Similar results were obtained for the dehalogenation of 4-iodobenzoic acid using a metal surface with and without a dielectric layer (Table S3, ESI†).
To summarise, non-thermal plasmas were shown to be viable initiators for radical chain reactions. Three reactions – dehalogenation of iodoarenes, 5-exo-trig cyclisation and trifluoromethylation with the Togni-II reagent – were carried out using two different plasma set-ups in the absence of other initiators. Excellent yields were recorded for all three reactions under optimised conditions. The short reaction times, mild conditions, scalability and a possibility to run solvent-free reactions make plasma initiation an attractive alternative to conventional redox, thermal or photochemical initiation. We note, however, that these are only preliminary results, and much further work is required to gain mechanistic understanding of how plasma properties, mass transfer and other reaction conditions affect the efficiency of the reactions.
This work was supported by the Leverhulme Trust (grant RPG-2013-079) and EPSRC (EP/K018388/1 and EP/H003797/2).
Notes and references
- X. Lu, G. V. Naidis, M. Laroussi, S. Reuter, D. B. Graves and K. Ostrikov, Phys. Rep., 2016, 630, 1 CrossRef CAS.
- (a) K. Bazaka, M. V. Jacob and K. Ostrikov, Chem. Rev., 2016, 116, 163 CrossRef CAS PubMed; (b) M. E. Ryan, A. M. Hynes and J. P. S. Badyal, Chem. Commun., 1996, 37 CAS.
- M. M. Hefny, C. Pattyn, P. Lukes and J. Benedikt, J. Phys. D: Appl. Phys., 2016, 49, 404002 CrossRef.
- Y. Gorbanev, N. Stehling, D. O'Connell and V. Chechik, Plasma Sources Sci. Technol., 2016, 25, 055017 CrossRef.
- A. Privat Maldonado, D. O'Connell, E. Welch, R. Vann and M. W. van der Woude, Sci. Rep., 2016, 6, 35646 CrossRef CAS PubMed.
- Y. Gorbanev, D. O'Connell and V. Chechik, Chem. – Eur. J., 2016, 22, 3496 CrossRef CAS PubMed.
- D. O. Jang, Tetrahedron Lett., 1996, 37, 5367 CrossRef CAS.
- (a) C. W. White, J. H. Jackson, A. Abuchowski, G. M. Kazo, R. F. Mimmack, E. M. Berger, B. A. Freeman, J. M. McCord and J. E. Repine, J. Appl. Physiol., 1989, 66, 584 CAS; (b) C. H. Liao, S. F. Kang and F. A. Wu, Chemosphere, 2001, 44, 1193 CrossRef CAS PubMed.
- Y. Kita, H. Nambu, N. G. Ramesh, G. Anilkumar and M. Matsugi, Org. Lett., 2001, 3, 1157 CrossRef CAS PubMed.
- Y. Gorbanev, R. Soriano, D. O'Connell and V. Chechik, J. Visualized Exp., 2016, 117, e54765 Search PubMed.
- A. Lindsay, C. Anderson, E. Slikboer, S. Shannon and D. Graves, J. Phys. D: Appl. Phys., 2015, 48, 424007 CrossRef.
- P. Rumbach, D. M. Bartels, R. M. Sankaran and D. B. Go, Nat. Commun., 2015, 6, 7248 CrossRef CAS PubMed.
- D. Leifert, D. G. Artiukhin, J. Neugebauer, A. Galstyan, C. A. Strassert and A. Studer, Chem. Commun., 2016, 52, 5997 RSC.
- B. Zhang, C. Mück-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792 CrossRef CAS PubMed.
- (a) I. Ryu, N. Sonoda and D. P. Curran, Chem. Rev., 1996, 96, 177 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC.
- (a) Q. Wang, X. Dong, T. Xiao and L. Zhou, Org. Lett., 2013, 15, 4846 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Org. Lett., 2014, 16, 1216 CrossRef CAS PubMed; (c) J. Lei, X. Wu and Q. Zhu, Org. Lett., 2015, 17, 2322 CrossRef CAS PubMed.
- S. Reuter, J. Winter, A. Schmidt-Bleker, D. Schroeder, H. Lange, N. Knake, V. Schulz-von der Gathen and K.-D. Weltmann, Plasma Sources Sci. Technol., 2012, 21, 024005 CrossRef.
- P. Rumbach, D. M. Bartels, R. M. Sankaran and D. B. Go, J. Phys. D: Appl. Phys., 2015, 48, 424001 CrossRef.
- J. Franclemont, X. Fan and S. M. Thagard, J. Phys. D: Appl. Phys., 2015, 48, 424004 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and further data. See DOI: 10.1039/c7cc01157a |
| ‡ The reaction times represent the overall exposure to plasma. To avoid significant temperature increase in the case of sinusoidal kHz plasma, each 1 min exposure was alternated with 1 min cooling time (no plasma, He flow only). |
| This journal is © The Royal Society of Chemistry 2017 |