
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnesium ion mobility in post-spinels accessible at ambient pressure†
Daniel C.
Hannah
 a,
Gopalakrishnan
Sai Gautam
ab,
Pieremanuele
Canepa
ab,
Ziqin
Rong
b and
Gerbrand
Ceder
*abc
a,
Gopalakrishnan
Sai Gautam
ab,
Pieremanuele
Canepa
ab,
Ziqin
Rong
b and
Gerbrand
Ceder
*abc
aMaterials Sciences Division, Lawrence Berkeley National Laboratory, CA 94270, USA. E-mail: gceder@berkeley.edu
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Materials Science and Engineering, University of California Berkeley, Berkeley, CA 94270, USA
First published on 18th April 2017
Abstract
We propose that Ti-containing post-spinels may offer a practically-accessible route to fast multivalent ion diffusion in close-packed oxide lattices, with the caveat that substantial thermodynamic driving forces for conversion reactions exist.
Magnesium batteries present an appealing means of improving upon the energy density of Li-ion batteries, due to the potential usage of a metal anode instead of the insertion structures common to Li-ion architectures,1,2,9 offering a major improvement in volumetric energy density (3833 A h L−1vs. 800 A h L−1 for Li+ in graphite).3 A major obstacle in the development of Mg batteries continues to be the poor electrochemical performance of Mg intercalation cathodes, stemming predominantly from the low mobility of Mg ions within highly ionic oxide frameworks.3–7 Mg mobility can often be improved by utilizing a sulfide cathode, with higher covalency and volume per anion reducing the barriers to Mg motion.8 However, increased mobility comes at the expense of reduced energy density, as sulfides generally intercalate Mg at lower voltages relative to oxides.9–13 As the search for fast-diffusing, high-energy density cathodes remains an active area of research,2,6 it remains to be seen whether fast Mg diffusion in a close-packed oxide framework is even possible in principle.
The post-spinel family of compounds presents a possible solution to this conundrum, albeit with complications arising from the fact that many post-spinel structures require synthesis conditions in the GPa pressure range.14,17 Utilizing first principles calculations of ionic mobility, Ling et al. demonstrated that CaFe2O4-type MgMn2O4 exhibits a relatively low barrier to Mg2+ migration, leading to post-spinels being proposed as Mg battery cathodes.15
The CaFe2O4-type Pnma structure, depicted graphically in Fig. 1a, is comprised of A-site (e.g. Ca) ions residing in eight-coordinate bicapped trigonal prism sites and distorted BO6 (B = transition metal) octahedra, two of which share an edge to form double octahedral units. The corner connections of four double octahedral units form 1D channels in the a direction, along which ionic diffusion is expected to occur.
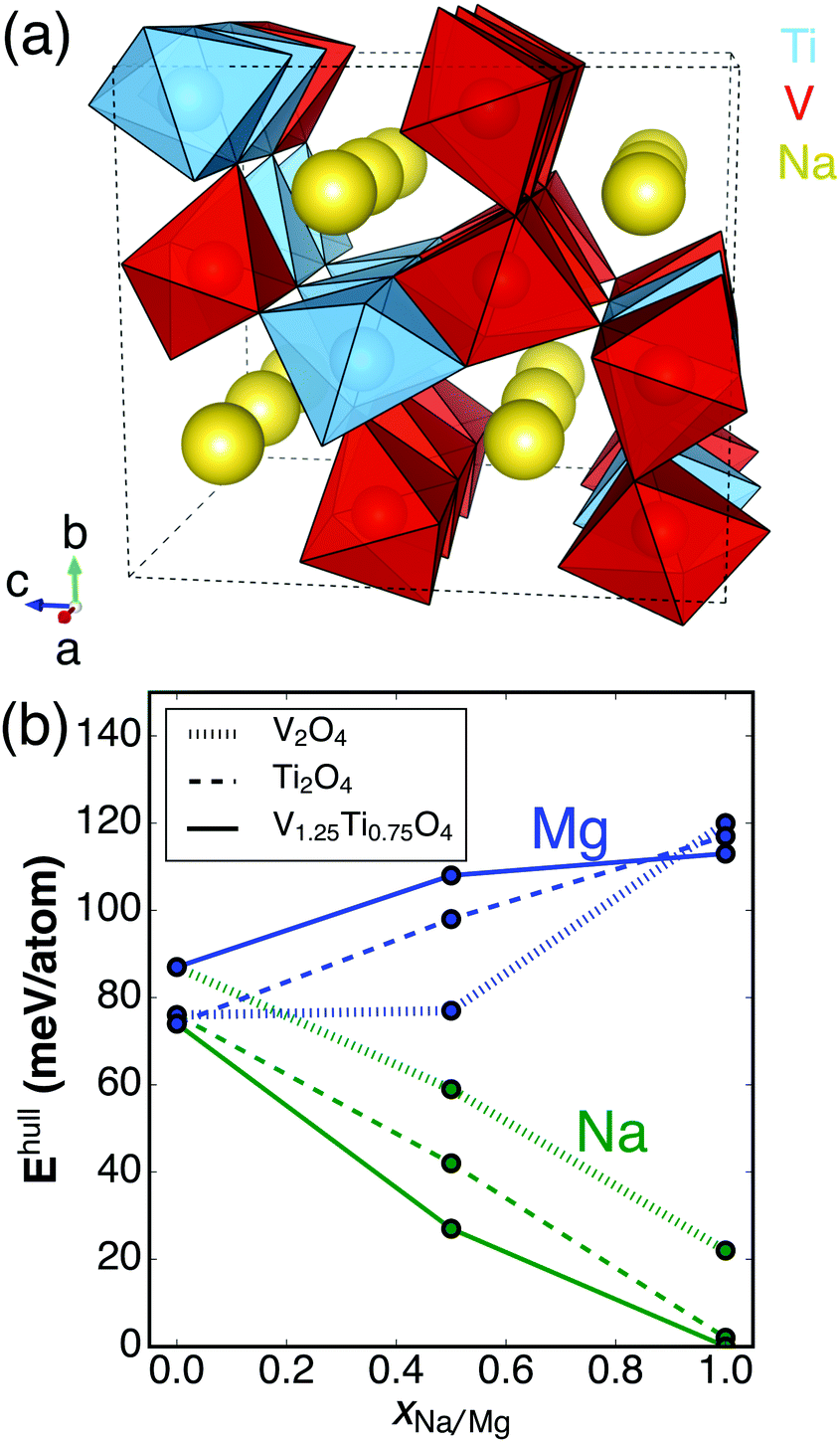 | ||
| Fig. 1 (a) The lowest-energy ordering of V and Ti atoms in a 3 × 1 × 1 supercell of Na4V5Ti3O16 based on a ranking of the DFT energy of possible orderings whose structures were generated using a previously published algorithm.18 (b) DFT-derived energies above the convex hull (Ehull) for AxV2−yTiyO4 (A = vacancy, Na, Mg) as a function of Na (green) or Mg (blue) concentration. Three different Ti concentrations are shown: y = 0 (dotted line), y = 0.75 (solid line), y = 2 (dashed line). The partially sodiated/magnesiated structures are the lowest-energy vacancy/(Na/Mg) orderings in the respective AxV2−yTiyO4 unit cells. | ||
The manganese oxide post-spinels have only been prepared at high pressures (>4 GPa),16,17 and this requirement for high pressures has, to date, stifled attempts to prepare sufficient quantities of magnesiated post-spinel compounds for electrochemical characterization. However, other transition metal (TM) oxides do not suffer from this limitation. For example, NaTi2O4 is stable at ambient pressure in the CaFe2O4-type structure.19 Because Mg intercalation into Ti hosts generally occurs at a low voltage,12 V might seem a more appealing transition metal from an energy density standpoint.7,12 However, NaV2O4 is only stable in the CaFe2O4-type structure at high pressure.20 Interestingly, it was recently demonstrated that substitution of a small amount of Ti (∼37.5%) into NaV2O4 stabilizes NaV1.25Ti0.75O4 at ambient pressure.21 If Na ions could be removed from the structure and replaced by Mg, the compound may exhibit fast Mg diffusion. To assess this possibility, we present here a first-principles study of Mg insertion in the V2−xTixO4 system with the CaFe2O4-type structure. As the structure is usually prepared with Na occupying the A-site (yellow spheres in Fig. 1a),19–21 we also present an analogous study of Na removal, which would be required prior to Mg insertion.
Our density functional theory (DFT)22-based calculations, as detailed in the ESI,† indicate that Mg insertion occurs in the range of 0.6–1.5 V (against Mg metal) depending on the V/Ti ratio, while Na is removed in the range of 1.5–2.5 V (against Na metal). The insertion of Na is generally accompanied by a modest volumetric expansion (predominantly along the b axis), while Mg insertion may result in expansion or contraction depending upon the V/Ti ratio. Finally, we find Na mobility to be acceptable (<650 meV migration barrier in most cases), with excellent Mg mobility (<300 meV migration barrier) expected. However, low barriers to Mg migration can be a result of the inherent instability of Mg in the V/Ti post-spinel lattice, and we predict a strong thermodynamic driving force for conversion reactions to occur, which would result in the decomposition of the post-spinel.
Fig. 1a displays the lowest-energy V/Ti ordering in the CaFe2O4-type crystal structure among all possible V/Ti orderings on a 3 × 1 × 1 supercell of Na4V5Ti3O16. Fig. 1b shows the calculated stability of each compound considered in this study, based on the Materials Project database.23 Specifically, the energy above the ground state hull (Ehull) is shown for AxV2−yTiyO4 (0 ≤ x ≤ 1, 0 ≤ y ≤ 2, A = Na, Mg, or vacancy). Ehull describes the amount of energy released by the decomposition of a compound into the most stable compound(s) at that chemical composition. Stable compounds have Ehull = 0 eV.23 A positive Ehull does not necessarily mean a material cannot be synthesized; metastable phases are often attainable through special synthetic routes. Indeed, metastable phases are frequently observed in cathode materials, including spinel-Mn2O428 obtained by delithiation of LiMn2O4 and ε-Mg0.5V2O5 observed on magnesiating α-V2O5.29 As a guideline, ∼80% of the compounds in the ICSD exhibit Ehull up to ∼40 meV per atom.24
From inspection of Fig. 1b, it is clear that sodiation stabilizes the post-spinel framework, whereas magnesiation destabilizes it. The fully Na-intercalated structures are predicted to be stable (Ehull < 2 meV per atom for NaTi2O4 and NaV1.25Ti0.75O4), while NaV2O4 is metastable (Ehull = 22 meV per atom). The stabilization of NaV1.25Ti0.75O4 with respect to NaV2O4 is consistent with the experimental finding that only the former compound is stable at ambient pressure.21 Two hypotheses exist to explain the stabilization mechanism of the NaV1.25Ti0.75O4 phase: (i) the substitution of Ti4+ for V4+ dilutes the frustrated magnetic network predicted to exist in NaV2O4,21,25 and (ii) the substitution of a larger B-site ion should make the CaFe2O4-type structure more favorable.14,26
The empty and magnesiated forms of the V/Ti post-spinels are considerably less stable (Ehull = 113, 117, 120 meV per atom for MgV2O4, MgV1.25Ti0.75O4, and MgTi2O4, respectively, and Ehull = 87, 77, 74 meV per atom for V2O4, V1.25Ti0.75O4, and Ti2O4, respectively, as shown in Fig. 1b). Some kinetic stabilization against phase transitions during cycling might be expected on the basis of poor B-site ion mobility15 and the requirement for substantial re-arrangement of the underlying oxygen lattice to occur (as would be the case, for example, in a post-spinel → spinel transition).27 In particular, if the empty structure can be obtained via desodiation, the V1.25Ti0.75O4 host material is as stable as the 50% magnesiated structure, indicating that some degree of reversible magnesiation may be feasible. Nevertheless, the computed values of Ehull suggest there is a substantial thermodynamic driving force for decomposition, especially for the fully magnesiated cathodes (see decomposition reactions in the ESI†). Decomposition reactions can be detrimental not only due to the destruction of cathode material, but also because many products (such as MgO) are passivating with regard to ionic conductivity and can prevent (de-)intercalation of remaining material.30
Fig. 2 reports the calculated properties of Na/Mg (de-)intercalation into the V2−xTixO4 system. Fig. 2a shows the variation of unit-cell volume upon Na or Mg insertion (y-axis) into the charged (“empty”) structure as a function of Ti concentration (x-axis). A non-negligible increase (∼4–6%) is noted for all three compounds upon Na insertion, with the largest increase observed for the mixed V/Ti system. Insertion of Mg yields a smaller increase (∼2%) for both V-containing compounds, but a decrease in volume (∼2%) for Ti2O4. For both Na and Mg insertion the pure Ti2O4 compound has the lowest volume change. This is consistent with the low volume change associated with Li intercalation in Li[Li1/3Ti5/3]O4 (LTO), attributed to the non-bonding nature of the t2g electron which is added when Ti4+ is reduced to Ti3+. Indeed, LTO is one of only two known zero-strain intercalation electrodes.31,32 Similarly, Fig. 2b and c show the variation of the a, b, and c unit cell parameters upon Na (Fig. 2b) or Mg (Fig. 2c) insertion (y-axis) as a function of Ti concentration (x-axis). From Fig. 2b, Na insertion into V-containing compounds produces minor changes in the a and c lattice parameters (≤2%) while yielding a substantial increase (∼4–5%) along b. The lattice parameter changes are generally smaller upon Na insertion into Ti2O4, with b increasing by ∼0.5% but a and c decreasing by ∼1%.
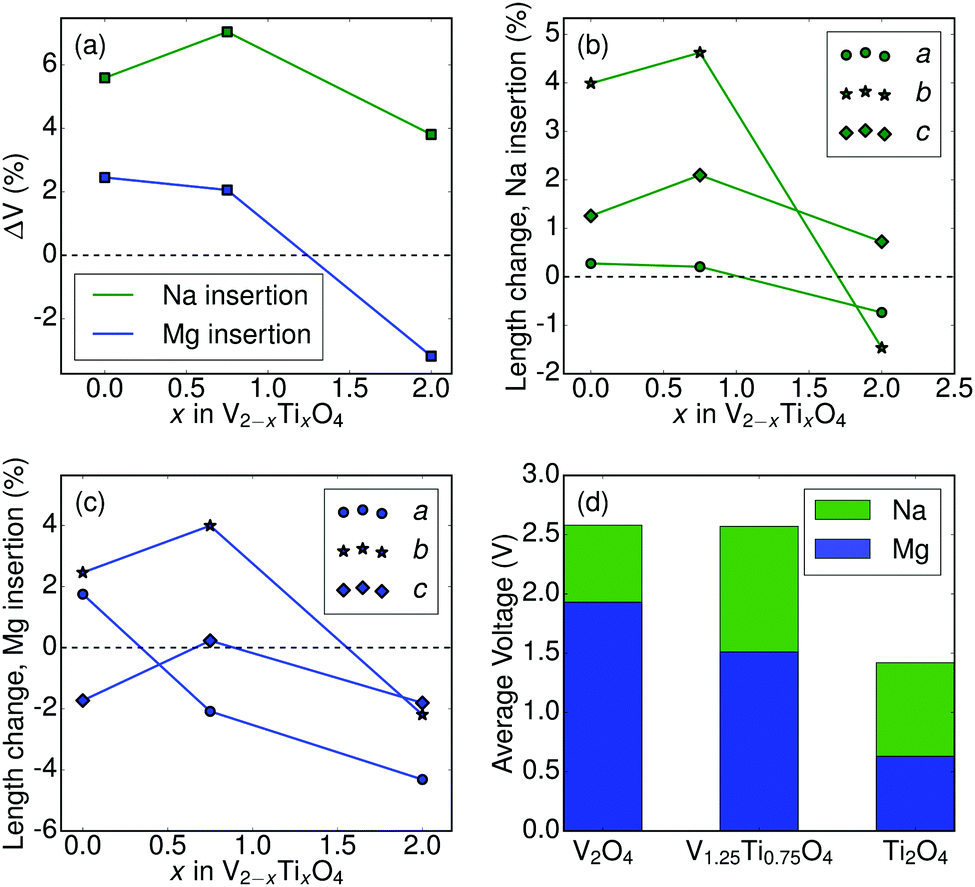 | ||
| Fig. 2 (a) Volume change upon ion (Na or Mg; indicated on the figure) insertion into V2−xTixO4. (b and c) Calculated changes in lattice parameters upon Na (b) or Mg (c) insertion into V2−xTixO4. Each lattice parameter (a, b, or c) is represented by a different shape as indicated in the legend. Axis labeling conventions match those in Fig. 1b. (d) Calculated average voltages for the (de-)intercalation of Na/Mg ions in CaFe2O4-type V2−xTixO4 (x = 0, 0.75, 2). The mobile ion (Na or Mg) is indicated by color on the plot. | ||
The variation of lattice parameters upon Mg insertion (Fig. 2c) differs somewhat from the Na insertion behavior displayed in Fig. 2b. In the V-containing compounds, the b parameter increases up to ∼4% (similar to Na insertion), with a and c parameters exhibiting minor variations. All three lattice parameters decrease upon Mg insertion into Ti2O4.
Fig. 2d displays the computed average voltage for Na/Mg (de-)intercalation in the 3 compounds considered in this study (see concentration-dependent voltage curves in ESI†). Note that voltage values for the V1.25Ti0.75O4 compound were calculated using the ground state ordering, as indicated in Fig. 1a. Voltages for Na (de-)intercalation (1.4–2.6 V) are within the range of voltages reported for other Na-ion cathodes and would not present an obstacle to electrochemical removal of Na.33 While the voltage of Mg insertion/removal in Ti2O4 is too low (0.63 V) to be of practical utility as a battery cathode, the V1.25Ti0.75 and V2O4 post-spinels exhibit average voltages in excess of 1.5 V. Considering theoretical capacities in the range of 281–285 mA h g−1, Mg insertion at 1.5 V would yield specific energies greater than the present state-of-the-art Chevrel and thiospinel cathodes (80–190 mA h g−1 at ∼1.1 V).9,10
Fig. 3 displays the DFT-calculated energy profiles (from the Nudged Elastic Band method,34 see ESI†) along the migration path of Na or Mg ions. The energy of a particular image (relative to the starting stable site) is displayed on the y-axis as a function of progress along the migration trajectory (x-axis). Energy profiles are displayed for a single-ion in the empty (“charged”) host as solid lines in Fig. 3 and for a single-vacancy in the filled (“discharged”) host as dashed lines. A comparison of the Na migration profiles (Fig. 3a, c and e) to the Mg ion profiles (Fig. 3b, d and f) reveals two primary differences. Firstly, migration barriers for Na as indicated by the maxima in the energy profile, (Em) in Fig. 3, are higher in the single ion limit (500–1000 meV) than in the single vacancy limit (∼400 meV), while the reverse is generally true for the Mg barriers (charged: ∼175 meV, discharged: 200–300 meV, with the V2O4 barriers being comparable for the charged and discharged limits). In the case of Na, the insertion of Na reduces the transition metal and lowers electrostatic repulsion in the lattice, lowering the barrier in the discharged state. The insertion of Mg is accompanied by an even greater reduction of the transition metal but also results in a volume reduction (Fig. 2a) of the post-spinel, yielding a higher barrier in the discharged state. Secondly, the shape of the migration profile is slightly different for Mg atoms, particularly for the Ti-containing compounds. This is probably related to a difference in stable sites: Mg occupies a different site in the channel as compared to Na, resulting in a different migration path (see ESI†). These differences become more pronounced as the Ti concentration increases owing to a concomitant increase in channel size. The overall barriers for Mg migration in all three structures are much lower than for Na. Magnesium migration barriers are likely lower because the magnesiated post-spinels are much less stable than the sodiated post-spinels (Fig. 1a), indicating a higher-energy starting site for the Mg migration path and thus reduced activation energy.6
 | ||
| Fig. 3 Activation barriers for ionic migration between neighboring sites in CaFe2O4-phase materials, as determined by DFT-coupled Nudged Elastic Band calculations. Barriers are shown for (a) Na in V2O4, (b) Mg in V2O4, (c) Na in V1.25Ti0.75O4, (d) Mg in V1.25Ti0.75O4, (e) Na in Ti2O4 and (f) Mg in Ti2O4. The solid lines correspond to the limit of a single diffusing ion per supercell (“charged” state), while the dashed lines correspond to the limit of a single Na/Mg vacancy diffusion (“discharged” state). | ||
The migration barriers (Em) in Fig. 3 can be used to provide an estimate of the ionic diffusivity. Using a simple random walk analysis and a vibrational frequency of 1012 s−1, an Em of ∼525 meV corresponds to a room-temperature ionic diffusivity of ∼10−12 cm2 s−1.6 Considering the relationship between diffusivity and particle size, migration barriers up to ∼650 meV yield reasonable diffusivity in 100 nm particulates, with larger particles necessitating lower migration barriers. As a result, the migration barriers presented in Fig. 3 are well within the range of acceptable values and indicate that the extraction of Na and subsequent insertion of Mg should be feasible from a kinetic standpoint.
While the diffusivity of Na and Mg in the V/Ti-compounds are predicted to be favorable, it is important to note that the thermodynamic driving force for decomposition (Fig. 1b) is large enough that the expected kinetic stabilization may not be sufficient to prevent conversion reactions (eqn (S1)–(S3), ESI†) from occurring. As the sodiated forms of the V/Ti and Ti post-spinels are stable at ambient pressure and Na mobility is sufficient to permit extraction, experimental testing of magnesiation of post-spinels should be possible.
To conclude, we have utilized first-principles calculations to assess, for the first time, the performance of post-spinel phase V and Ti oxides as cathodes for magnesium batteries. Because the preparation of these compounds would likely require a sodiated starting material, we have also studied the thermodynamics and kinetics of these compounds with sodium. Consistent with previous reports on manganese oxide post-spinels, Na+ and Mg2+ migration generally exhibit a sufficiently low energetic barrier for extraction/intercalation to be feasible. Importantly, these materials differ from the manganese oxide post-spinels in that NaV1.25Ti0.75O4 and NaTi2O4 are stable at ambient pressure, providing a much easier route to synthesis. While the post-spinel lattice is expected to be kinetically stabilized, the thermodynamic instability (Ehull > 100 meV per atom) suggests that experimental efforts to intercalate Mg into V2−xTixO4 should be monitored carefully for the occurrence of parallel conversion reactions leading to the decomposition of the post-spinel structures. As the requirement for high pressures in the GPa range has so-far been a major obstacle to the experimental investigation of magnesium mobility in the post-spinel framework, the results presented here suggest that the V and Ti oxide post-spinels may represent a means of achieving the milestone of high Mg-mobility.
The current work is fully supported by the Joint Center for Energy Storage Research (JCESR), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science and Basic Energy Sciences. This study is supported by Subcontract 3F-31144. The authors thank the National Energy Research Scientific Computing Center (NERSC) for computing resources. The authors would also like to thank Jordi Cabana and Linda Nazar for helpful discussions regarding the synthesis of post-spinel.
References
- D. Aurbach, Y. Cohen and M. Moshkovich, Electrochem. Solid-State Lett., 2001, 4, A113–A116 CrossRef CAS.
- P. Canepa, G. Gautam, D. Hannah, R. Malik, M. Liu, K. Gallagher, K. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 CAS.
- G. Gershinsky, H. D. Yoo, Y. Gofer and D. Aurbach, Langmuir, 2013, 29, 10964–10972 CrossRef CAS PubMed.
- G. Amatucci, F. Badway, A. Singhal, B. Beaudoin, G. Skandan, T. Bowmer, I. Plitz, N. Pereira, T. Chapman and R. Jaworski, J. Electrochem. Soc., 2001, 148, A940–A950 CrossRef CAS.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- G. S. Gautam, P. Canepa, R. Malik, M. Liu, K. Persson and G. Ceder, Chem. Commun., 2015, 51, 13619–13622 RSC.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026–1031 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, Energy Environ. Sci., 2016, 9, 2273–2277 CAS.
- M. Liu, A. Jain, Z. Rong, X. Qu, P. Canepa, R. Malik, G. Ceder and K. A. Persson, Energy Environ. Sci., 2016, 9, 3201–3209 CAS.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974 CAS.
- A. Emly and A. Van der Ven, Inorg. Chem., 2015, 54, 4394–4402 CrossRef CAS PubMed.
- H. Müller-Buschbaum, J. Alloys Compd., 2003, 349, 49–104 CrossRef.
- C. Ling and F. Mizuno, Chem. Mater., 2013, 25, 3062–3071 CrossRef CAS.
- X. Liu, X. Wang, A. Iyo, H. Yu and H. Zhou, et al. , J. Mater. Chem. A, 2014, 2, 14822–14826 CAS.
- K. Yamaura, Q. Huang, L. Zhang, K. Takada, Y. Baba, T. Nagai, Y. Matsui, K. Kosuda and E. Takayama-Muromachi, J. Am. Chem. Soc., 2006, 128, 9448–9456 CrossRef CAS PubMed.
- G. L. Hart and R. W. Forcade, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 224115 CrossRef.
- J. Akimoto and H. Takei, J. Solid State Chem., 1989, 79, 212–217 CrossRef CAS.
- K. Yamaura, M. Arai, A. Sato, A. Karki, D. Young, R. Movshovich, S. Okamoto, D. Mandrus and E. Takayama-Muromachi, Phys. Rev. Lett., 2007, 99, 196601 CrossRef CAS PubMed.
- L. Viciu, A. Ryser, C. Mensing and J.-W. G. Bos, Inorg. Chem., 2015, 54, 7264–7271 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 Search PubMed.
- H. Sakurai, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 094410 CrossRef.
- O. Muller and R. Roy, The Major Ternary Structural Families, Springer, 1974 Search PubMed.
- Á. M. Arévalo-López, A. J. Dos santos García, E. Castillo-Martínez, A. Durán and M. Á. Alario-Franco, Inorg. Chem., 2010, 49, 2827–2833 CrossRef PubMed.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142–147 CrossRef CAS.
- A. Mukherjee, N. Sa, P. J. Phillips, A. Burrell, J. Vaughey and R. F. Klie, Chem. Mater., 2017, 29, 2218–2226 CrossRef CAS.
- H. Wang, P. Senguttuvan, D. L. Proffit, B. Pan, C. Liao, A. K. Burrell, J. T. Vaughey and B. Key, ECS Electrochem. Lett., 2015, 4, A90–A93 CrossRef CAS.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431–1435 CrossRef CAS.
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 CAS.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the methods used in this work provided. See DOI: 10.1039/c7cc01092c |
| This journal is © The Royal Society of Chemistry 2017 |