
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling transmembrane protein concentration and orientation in supported lipid bilayers†
P.
Bao
 a,
M. L.
Cartron
b,
K. H.
Sheikh
c,
B. R. G.
Johnson
a,
C. N.
Hunter
b and
S. D.
Evans
*a
a,
M. L.
Cartron
b,
K. H.
Sheikh
c,
B. R. G.
Johnson
a,
C. N.
Hunter
b and
S. D.
Evans
*a
aSchool of Physics & Astronomy, University of Leeds, LS2 9JT, UK. E-mail: s.d.evans@leeds.ac.uk
bDepartment of Molecular Biology & Biotechnology, University of Sheffield, S10 2TH, UK
cSchool of Biomedical Science, University of Leeds, LS2 9JT, UK
First published on 31st March 2017
Abstract
The trans-membrane protein – proteorhodopsin (pR) has been incorporated into supported lipid bilayers (SLB). In-plane electric fields have been used to manipulate the orientation and concentration of these proteins, within the SLB, through electrophoresis leading to a 25-fold increase concentration of pR.
The cell membrane defines the boundary between the cell interior and the extracellular environment and acts as an impermeable barrier to the transfer of ions and small molecules and thus allows the generation of electrical and chemical gradients. Further, it acts as host to a wide variety of membrane proteins that regulate a range of cellular processes and represent the targets of more than 60% of modern pharmaceuticals.1 Membrane proteins are difficult to study because of their inherent instability outside of their membranous environment and this has led to a variety of approaches being investigated, including the use of lipid discs; droplet interface bilayers; and solid supported, or tethered, lipid bilayer (SLBs).2–6 The latter have potential for the study of membrane protein function, peptide–membrane interactions and for understanding the role of phase separation and domains in lipid membranes.7–10 Typically, SLBs are formed via vesicle adsorption, rupture and fusion at the surface.11 This process is effective for lipid-only vesicles and vesicles with low protein content. Numerous studies have demonstrated the incorporation of functional membrane proteins into such bilayers and those involved in the photosynthetic process have recently been reviewed.12 However, at higher protein concentrations vesicles tend to remain intact thereby limiting the amount of protein that can be incorporated within the SLB.13,14 It is therefore of interest to develop methods to increase membrane protein concentration, and orientation, post bilayer formation.5,15–22 Approaches followed to date include the use of hydrodynamic drag, which relies on the frictional interaction between a fluid flowing across the membrane and protruding membrane components.15,23 Jönsson et al., have used this to demonstrate the accumulation and separation of membrane associated proteins, streptavidin and cholera toxin, coupled to receptors in lipid membrane.15 Alternatively, electric fields can be used to induce electrophoretic and electro-osmotic manipulation of charged species.13,19,24–27 Pace et al. recently demonstrated the successful separation of four membrane-associated species according to their different size/charge ratio.19 Further, we have shown the possibility of moving and concentrating an integral membrane protein, Cyma, which has a single membrane spanning α-helix.17 However, until now, there have been no reports on the electric-field based manipulation of larger integral membrane proteins within SLBs.
The light-activated proton pump, proteorhodopsin (pR), has seven membrane spanning domains and charged hydrophilic loops extending beyond the membrane with a molecular weight of ∼29 kDa. It can be expressed in E. coli and has broad potential application from photovoltaic to data storage.28–30 Here we show that pRs reconstituted into lipid bilayers are mobile and can be concentrated via the application of in-plane electric field (Fig. 1).
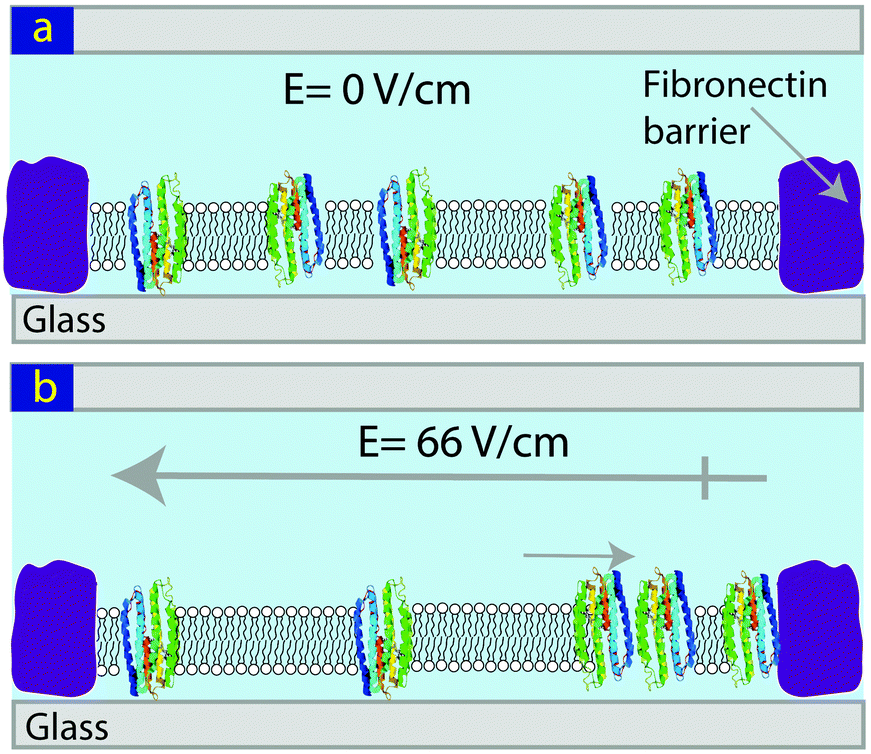 | ||
| Fig. 1 Schematic showing the concentration of pR in a SLB using an in-plane electric field. (a) Before, and (b) following the application of an E-field. | ||
SLBs were formed from ∼50 nm diameter vesicles of DOPC, containing 1 wt% Alexa488 labelled pR.31 Bilayer formation was evaluated using fluorescence microscopy and AFM. Fluorescence recovery after photo-bleaching (FRAP) data for Alexa488 labelled protein (pR-Alexa488) shows that the pR is mobile with a diffusion coefficient (D) of 0.39 ± 0.03 μm2 s−1 and the mobile fraction 87 ± 5% (Fig. S1, ESI†). This value for D is lower than the 1 to 4 μm2 s−1 typically observed for lipids in SLBs formed on glass but similar to observations for other membrane proteins in SLBs.32,33 For example haemolysin (Mw = 33 kDa) has been observed diffuse 3-fold slower than lipids in the SLBs on glass.33 Since the pR has an extra-membranous region that extends beyond the surface of bilayer by nearly 1 nm interaction with the underlying glass substrate could also contribute to reduced diffusivity and mobile fraction values.34–36
The force–distance curve obtained by AFM of pR-containing lipid bilayers on glass (Fig. 2a) show typical a “punch-through” profile found for SLBs and indicates a bilayer thickness of ∼5.6 ± 0.2 nm (n = 7).37 A particle density of ∼12 ± 9 per μm2 (n = 19), was observed when imaging SLBs on glass (Fig. 2b). Since the AFM scan speeds were only up to 10 μm s−1 this does not allow the observation of the mobile fraction of proteins and these particles are likely to be immobile pRs oligomers, as shown in Fig. S4c (ESI†). Control experiments on pure DOPC lipid bilayers on glass didn’t show any particles, Fig. S3 (ESI†). However, they did show the presence of some nanosized holes (25 ± 3 nm in diameter, n = 20) in both the case of pR-containing, and pure, lipid bilayers on glass (Fig. 2b and Fig. S2, S3, ESI†). These holes are not found in bilayers formed on mica (Fig. S4 and S5, ESI†). AFM imaging of the glass coverslips (no. 1 Menzel Co., Germany) indicated that these were in fact due to the defects in glass substrate (Fig. S3, ESI†). AFM imaging of pRs in the SLB on mica showed a greater proportion of immobile particles than that on glass (Fig. 2c and Fig. S4, ESI†), presumably due to increased interaction with the mica. The pRs protrude from the bilayer by around 1 nm (Fig. 2d). From fitting the height data we observe two populations of pRs that protrude on average by ∼0.8 and 1.3 nm (Fig. 2e). These values are close to the 0.9 nm reported by Klyszejko et al. for pR crystals.38 The different the heights likely reflect the two possible orientations of the pRs in SLBs and is supported by the similar percentages of the two populations (49% and 51%, respectively), which is expected from a nearly random formation of SLBs via vesicle rupture. Single particle tracking (SPT) of these proteins in SLBs on mica suggested pRs are immobile (Fig. S6, ESI†), therefore, the density of pRs on mica should roughly represent the density of pRs in the proteoliposome, as well as in the bilayer on glass. Particle analysis suggests the density of pR in the lipid bilayer is 50 ± 10 μm−2 (n = 10). From the initial concentration we would expect to observe ∼350 pR molecules per μm2 suggesting that the particles observed consist of oligomers of protein (predominantly hexamers). This is consistent with the earlier report of oligomer states of pR in the membrane of E. coli.39
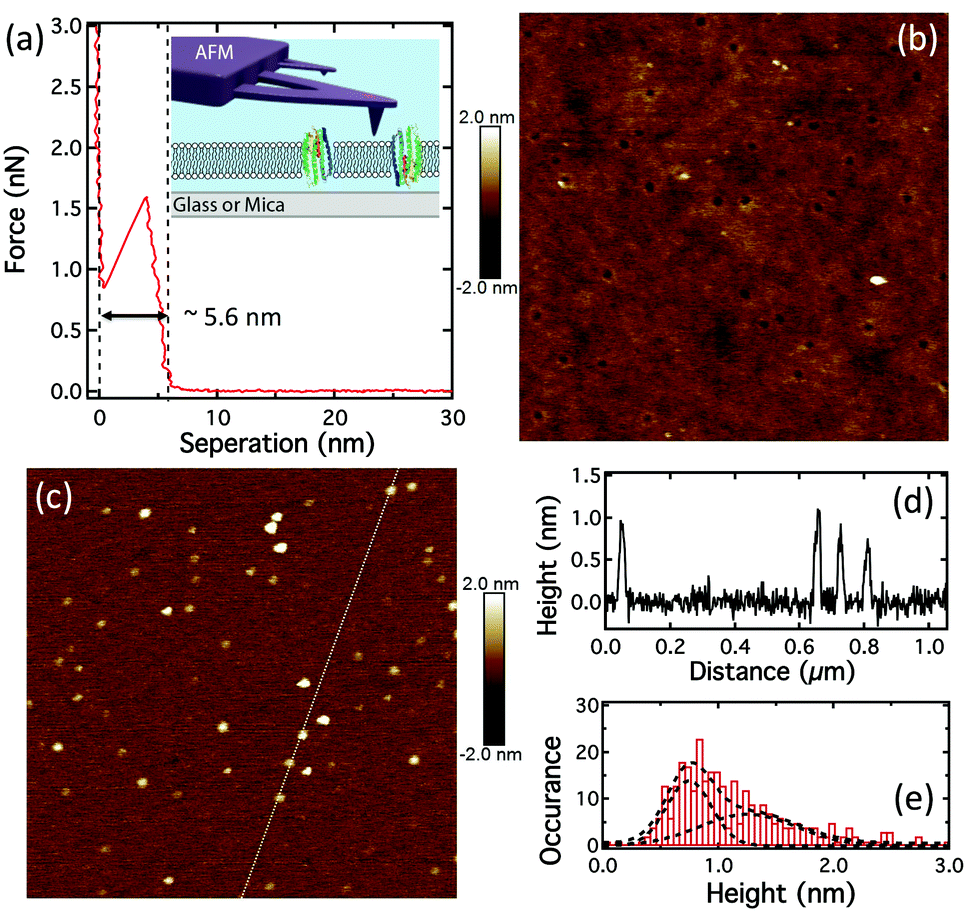 | ||
| Fig. 2 pR-Containing SLB formation. (a) The force-separation curve showing “punch-through” to determine bilayer thickness; inset: schematic of the AFM on a SLB. (b) AFM height image of pRs in SLB on glass (1 μm scan). (c) AFM height image of pRs in SLB on mica (1 μm scan). (d) The height profile along the dotted line in (c). (e) The histogram of the pR height, on mica, with fits to two peaks located at 0.8 and 1.3 nm respectively. | ||
To demonstrate the possibility of controlling the concentration of transmembrane proteins, bilayers containing 1 wt% pR-Alexa488 were formed by vesicle fusion of 50 nm proteoliposomes onto non-fibronectin covered regions of the surface.17 The fluorescence images of the patterned lipid bilayer for pR-Alexa488 are shown in Fig. 3. During the application of an electric field, 66 V cm−1, the pR-Alexa488 within the SLB moved toward the positive electrode and built-up as a function of time, as shown in Fig. 3a and b. These show that most of pR-Alexa488 molecules are concentrated in the trap regions. By increasing the length, L, of the bilayer structures we are able to systematically control the concentration of pR. The average fluorescence intensity for pR-Alexa488 in the trap head region (defined by white dotted line) for the longest device showed a ∼7-fold increase in intensity after application of the E-field. This value is lower than the maximum possible value of ‘×18’ estimated from the area ratio between the whole trap and the head region for corral 6 and is possibly due to lower pR mobility in increasingly crowded local environments, as indicated by the accumulation curves shown in Fig. 3c.
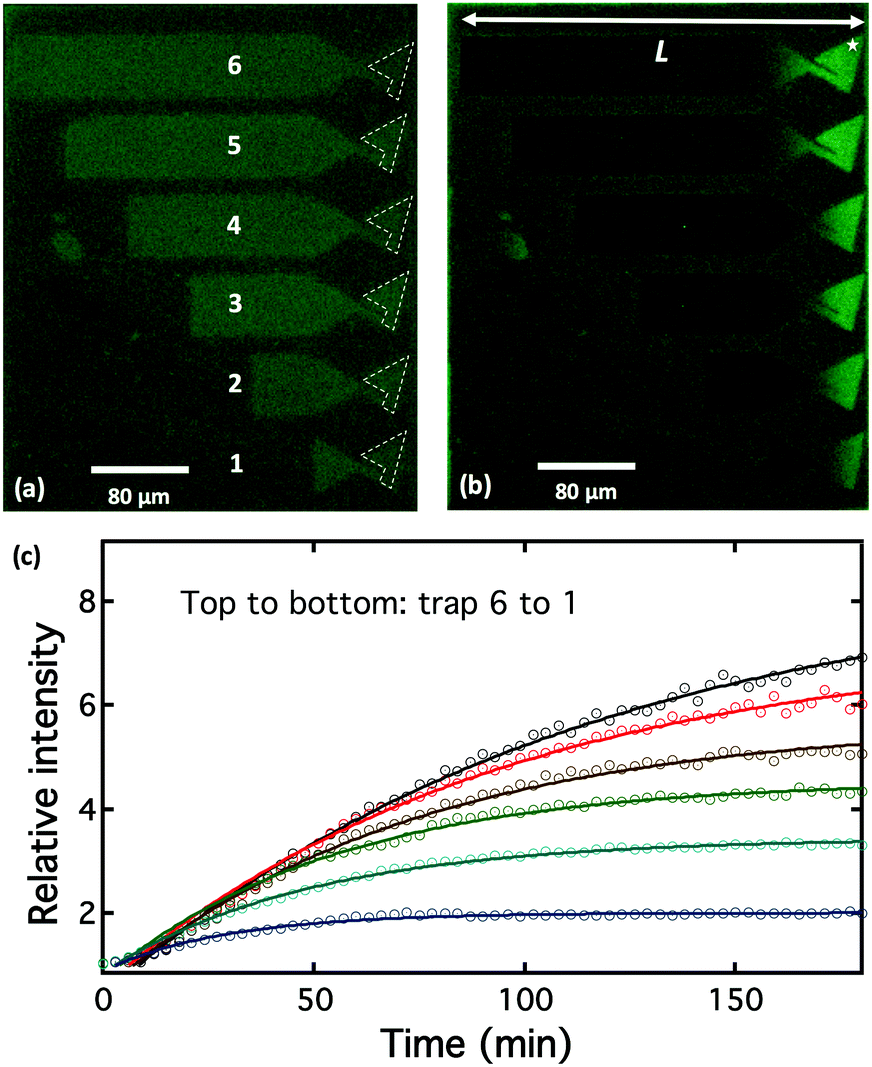 | ||
| Fig. 3 Electrophoresis of pR-Alexa488 in the DOPC lipid bilayer (brighter color regions) in “trap” structures of different lengths (labeled 1–6) formed by microcontact-printing fibronectin (darker color region) on a glass substrate. False-color fluorescence images of pR-Alexa488 using a FITC filter: (a) before application of E-field; (b) after E-field applied for180 min; (c) accumulation curves of pR-Alexa488 in the trap heads (regions defined by dashed lines in (a)) as a function of time. The star in (b) indicates where the AFM images were taken following the application of E-field. | ||
The build-up of pR was followed by AFM of the bilayer after electrophoresis, Fig. 4a. The density of pR in the head of the longest trap (labeled by an asterisk in the corral 6 in Fig. 3b) was ∼1264 ± 76 μm−2 after electrophoresis (Fig. 4a) compared to a density of ∼50 μm−2 prior to the application of the field (Fig. 2c) indicating a 25 fold build-up of pR concentration. We note that comparison between AFM and fluorescence is not straight forward as the AFM measures a local concentration within the trap and we expect an exponential profile in the protein distribution against the end of the barrier. Notwithstanding this, the AFM indicates that a significantly increased concentration of pRs can be achieved. It should be noted that with AFM we will likely only be “seeing” the immobile or low mobility proteins. This suggests that the mobility of pRs in the trap head-region is greatly reduced after concentration, which may be due to crowding or to oligomer/cluster formation of pRs.40 Many of the dots in AFM image appear triangular (n > 80) or hexagonal (n > 105), Fig. 4b, and might indicate the occurrence of clusters of pR oligomers. This emergence of clustering, at high protein concentration, may be indicative that the proteins retain their structural form and are not adversely perturbed during the in-plane electrophoresis. The electric field experienced by the pRs during our experiments (∼66 V cm−1) is several orders of magnitude below those typically experienced in their native environment (∼105 V cm−1). The height data of particles suggests two populations of pRs that protrude on average by ∼0.9 and 1.4 nm (Fig. 4c). These would be consistent with two orientations of pR as mentioned above. Following the electrophoretic concentration in the trap region we find that the ratio between these populations changes from (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (before E-field) to the (1:9) (after E-field), with the proteins presenting higher protrusions being the dominant population (89%). This suggests that pRs with different orientations have different mobility and that the pRs with higher protrusions move more readily in SLBs. Therefore, our approach could also be potentially very useful for the separation of the membrane proteins with different orientations. This is important as, so far, little has been achieved in the controlling the orientations of membrane proteins during or after reconstitution process.41,42
:1) (before E-field) to the (1:9) (after E-field), with the proteins presenting higher protrusions being the dominant population (89%). This suggests that pRs with different orientations have different mobility and that the pRs with higher protrusions move more readily in SLBs. Therefore, our approach could also be potentially very useful for the separation of the membrane proteins with different orientations. This is important as, so far, little has been achieved in the controlling the orientations of membrane proteins during or after reconstitution process.41,42
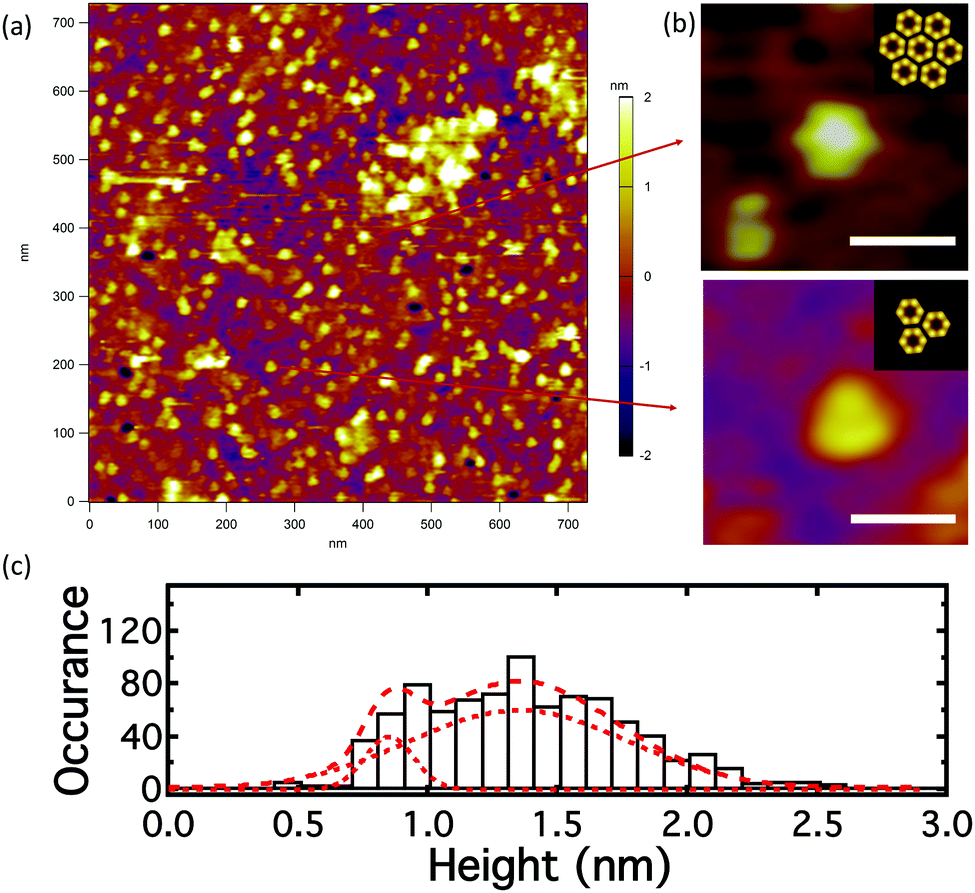 | ||
| Fig. 4 AFM image of pR-Alexa488 in the DOPC lipid bilayer, at the trap head-region in the longest trap (trap 6, Fig. 3), after the application of an E-field of 180 min. (a) Topology; (b) formation of regular oligomer clusters – hexagonal (n > 105), triangular (n > 80) structures; insets: schematics show the clustering state of pR oligomer (to the scale);38 (c) histogram of center height of particles in the AFM image. (Scale bar is 20 nm in (b)). | ||
In conclusion, this work demonstrates that integral membrane proteins, such as proteorhodopsin (Mw ∼ 29 kDa) with multiple trans-membrane spanning domains can not only be incorporated into SLBs but that they are able to diffuse freely. Further, under the application of an electric field these proteins move via electrophoresis and by patterning the SLBs into rectangular corrals of different lengths we have been able to demonstrate the controlled increase in protein concentration, following the application of the electric field. The protein concentrations were increased by over seven times, as determined by fluorescence and AFM. The approach is scalable and should permit significantly higher concentrations to be achieved by increasing the size of the trap region thereby opening the possibility of 2D crystallisation of membrane proteins within such bilayers. It also suggests that electrophoretic separation of integral membrane proteins, whilst still in their membranous environment should be feasible, at least for some membrane proteins. Furthermore, the change in the populations of the two different heights of pRs suggests our method could be used to separate membrane proteins according their orientation post SLB formation.
The authors would like to acknowledge the EPSRC (EP/ I012060/1), the Wellcome Trust ISSF, and NIHR-HTC for Colorectal Therapies for financial support. We also acknowledge Dr George Heath and Dr Johannes R. Roth for helpful discussions.
References
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- T. H. Bayburt and S. G. Sligar, FEBS Lett., 2010, 584, 1721–1727 CrossRef CAS PubMed.
- H. Bayley, B. Cronin, A. Heron, M. A. Holden, W. Hwang, R. Syeda, J. Thompson and M. Wallace, Mol. BioSyst., 2008, 4, 1191–1208 RSC.
- C. Yoshina-Ishii and S. G. Boxer, J. Am. Chem. Soc., 2003, 125, 3696–3697 CrossRef CAS PubMed.
- M. Stelzle, R. Miehlich and E. Sackmann, Biophys. J., 1992, 63, 1346–1354 CrossRef CAS PubMed.
- W. Knoll, C. W. Frank, C. Heibel, R. Naumann, A. Offenhäusser, J. Rühe, E. K. Schmidt, W. W. Shen and A. Sinner, Rev. Mol. Biotechnol., 2000, 74, 137–158 CrossRef CAS PubMed.
- L. K. Tamm and H. M. McConnell, Biophys. J., 1985, 47, 105 CrossRef CAS PubMed.
- A. Sumino, T. Dewa, M. Kondo, T. Mori, H. Hashimoto, A. T. Gardiner, R. J. Cogdell and M. Nango, Langmuir, 2011, 27, 1092 CrossRef CAS PubMed.
- H. Sato and J. B. Feix, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1245–1256 CrossRef CAS PubMed.
- S. D. Connell, G. Heath, P. D. Olmsted and A. Kisil, Faraday Discuss., 2013, 161, 91–111 RSC.
- Z. V. Leonenko, a. Carnini and D. T. Cramb, Biochim. Biophys. Acta, 2000, 1509, 131–147 CrossRef CAS.
- L. Wang, J. S. Roth, X. Han and S. D. Evans, Small, 2015, 11, 3306–3318 CrossRef CAS PubMed.
- H. Pace, L. Simonsson Nyström, A. Gunnarsson, E. Eck, C. Monson, S. Geschwindner, A. Snijder and F. Höök, Anal. Chem., 2015, 87, 9194–9203 CrossRef CAS PubMed.
- C. E. Dodd, B. R. G. Johnson, L. J. C. Jeuken, T. D. H. Bugg, R. J. Bushby and S. D. Evans, Biointerphases, 2008, 3, FA59–FA67 CrossRef CAS PubMed.
- P. Jönsson, A. Gunnarsson and F. Höök, Anal. Chem., 2011, 83, 604–611 CrossRef PubMed.
- A. van Oudenaarden and S. G. Boxer, Science, 1999, 285, 1046–1048 CrossRef CAS PubMed.
- M. R. Cheetham, J. P. Bramble, D. G. G. McMillan, L. Krzeminski, X. Han, B. R. G. Johnson, R. J. Bushby, P. D. Olmsted, L. J. C. Jeuken, S. J. Marritt, J. N. Butt and S. D. Evans, J. Am. Chem. Soc., 2011, 133, 6521–6524 CrossRef CAS PubMed.
- X. Han, M. R. Cheetham, K. Sheikh, P. D. Olmsted, R. J. Bushby and S. D. Evans, Integr. Biol., 2009, 1, 205 RSC.
- H. P. Pace, S. D. Sherrod, C. F. Monson, D. H. Russell and P. S. Cremer, Anal. Chem., 2013, 85, 6047–6052 CrossRef CAS PubMed.
- T. Motegi, H. Nabika and K. Murakoshi, Langmuir, 2012, 28, 6656 CrossRef CAS PubMed.
- P. Jonsson, J. McColl, R. W. Clarke, V. P. Ostanin, B. Jonsson and D. Kleneman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10328 CrossRef CAS PubMed.
- M. F. Poyton and P. S. Cremer, Anal. Chem., 2013, 85, 10803–10811 CrossRef CAS PubMed.
- B. Johansson, T. Olsson, P. Jonsson and F. Hook, Soft Matter, 2013, 9, 9414–9419 RSC.
- J. T. Groves and S. G. Boxer, Biophys. J., 1995, 69, 1972–1975 CrossRef CAS PubMed.
- M. Tanaka, J. Hermann, I. Haase, M. Fischer and S. G. Boxer, Langmuir, 2007, 23, 5638–5644 CrossRef CAS PubMed.
- D. J. Olson, J. M. Johnson, P. D. Patel, E. S. G. Shaqfeh, S. G. Boxer and G. G. Fuller, Langmuir, 2001, 17, 7396–7401 CrossRef CAS.
- J. van Weerd, S. O. Krabbenborg, J. Eijkel, M. Karperien, J. Huskens and P. Jonkheijm, J. Am. Chem. Soc., 2014, 136, 100–103 CrossRef CAS PubMed.
- P. Bertoncello, D. Nicolini, C. Paternolli, V. Bavastrello and C. Nicolini, IEEE Trans. Nanobiosci., 2003, 2, 124 CrossRef PubMed.
- R. B. Jensen, B. R. Kelemen, J. C. McAuliffer and W. C. Smith, US Pat., vol. 20040223323 A1, 2004 Search PubMed.
- C. Brauchle, N. Hampp and D. Oesterhelt, Adv. Mater., 1991, 3, 420 CrossRef.
- M. J. Hope, M. B. Bally, G. Webb and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1985, 812, 55–65 CrossRef CAS.
- S. Türkcan, M. U. Richly, A. Alexandrou and J.-B. Masson, PLoS One, 2013, 8, e53073 Search PubMed.
- F. Harb, J. Sarkis, N. Ferte and B. Tinland, Eur. Phys. J. E: Soft Matter Biol. Phys., 2012, 35, 1–9 CrossRef PubMed.
- G. Wilhelmina de Groot, S. Demarche, M. G. Santonicola, L. Tiefenauer and G. J. Vancso, Nanoscale, 2014, 6, 2228–2237 RSC.
- L. K. T. Michael and L. Wagner, Biophys. J., 2000, 79, 1400 CrossRef.
- E. A. Smith, J. W. Coym, S. M. Cowell, T. Tokimoto, V. J. Hruby, H. I. Yamamura and M. J. Wirth, Langmuir, 2005, 21, 9644–9650 CrossRef CAS PubMed.
- S. J. Attwood, Y. Choi and Z. Leonenko, Int. J. Mol. Sci., 2013, 14, 3514–3539 CrossRef CAS PubMed.
- A. L. Klyszejko, S. Shastri, S. A. Mari, H. Grubmüller, D. J. Muller and C. Glaubitz, J. Mol. Biol., 2008, 376, 35–41 CrossRef CAS PubMed.
- S. Hussain, M. Kinnebrew, N. S. Schonenbach, E. Aye and S. Han, J. Mol. Biol., 2015, 427, 1278–1290 CrossRef CAS PubMed.
- S. J. Bussell, D. L. Koch and D. A. Hammer, Biophys. J., 1995, 68, 1836–1849 CrossRef CAS PubMed.
- H. Liang, G. Whited, C. Nguyen and G. D. Stucky, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8212–8217 CrossRef CAS PubMed.
- R. Tunuguntla, M. Bangar, K. Kim, P. Stroeve, Caroline M. Ajo-Franklin and A. Noy, Biophys. J., 2013, 105, 1388–1396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FRAP, AFM results for bilayer on glass and mica, and experiment details. See DOI: 10.1039/c7cc01023k |
| This journal is © The Royal Society of Chemistry 2017 |