
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A highly stable sodium solid-state electrolyte based on a dodeca/deca-borate equimolar mixture†
L.
Duchêne
 ab,
R.-S.
Kühnel
a,
D.
Rentsch
a,
A.
Remhof
*a,
H.
Hagemann
b and
C.
Battaglia
a
ab,
R.-S.
Kühnel
a,
D.
Rentsch
a,
A.
Remhof
*a,
H.
Hagemann
b and
C.
Battaglia
a
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, CH-8600 Dübendorf, Switzerland. E-mail: arndt.remhof@empa.ch
bDépartement de Chimie-Physique, Université de Genève, CH-1211 Geneva 4, Switzerland
First published on 22nd March 2017
Abstract
Na2(B12H12)0.5(B10H10)0.5, a new solid-state sodium electrolyte is shown to offer high Na+ conductivity of 0.9 mS cm−1 at 20 °C, excellent thermal stability up to 300 °C, and a large electrochemical stability window of 3 V including stability towards sodium metal anodes, all essential prerequisites for a stable room-temperature 3 V all-solid-state sodium-ion battery.
All-solid-state sodium-ion batteries promise to simultaneously yield better performance and lower cost as compared to state-of-the-art lithium-ion technology using organic liquid electrolytes.1,2 To build such a competitive all-solid-state battery requires a solid-state electrolyte (SSE) with high ionic conductivity near room temperature and high thermal and electrochemical stability. Meeting these three requirements simultaneously still represents a major challenge. High ionic conductivity of or above 10−3 S cm−1 has been reached in oxide ceramics or more recently in sulfide phases but these materials still suffer from high electrode–electrolyte interface resistance or intrinsically small electrochemical stability windows of 1 V or less, respectively.3–10 Recently, complex hydride materials and particularly metal closo-borates have emerged as promising alternative SSEs.11,12 Metal closo-borates are salts with polyhedral BnHn2− (n = 6–12) anions, among which the salts of the B10H102− and B12H122− anions were early noted due to their high thermal and chemical stability.13 LiCB11H12 as a halogen free electrolyte was also predicted by theory14 and used in experiments to synthesize a Mg-electrolyte.15closo-Borate and related carborate salts (with monovalent CBn−1Hn− anions) also exhibit structural transitions to highly conductive phases, above room temperature.16–19 Strategies to stabilize these superionic phases at room temperature and below, including mechanical milling and cation or anion substitutions, have been implemented.20–23 Yet, studies on closo-borates have focused almost exclusively on improving conductivity, while only preliminary work has been performed on the assessment of their electrochemical stability window and of their suitability for integration into a battery. Here, we report a new sodium superionic conductor Na2(B12H12)0.5(B10H10)0.5 with a conductivity close to 10−3 S cm−1 at room temperature (20 °C). We further demonstrate that this material is compatible with a sodium metal anode and fulfils all prerequisites to be used in a 3 V all-solid-state sodium-ion battery.
Fig. 1a shows differential scanning calorimetry (DSC) data for the individual precursors Na2B12H12 and Na2B10H10 and their reacted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equimolar mixture. Both precursors show a characteristic endothermic peak at the temperature of their respective structural phase transition, as previously described.24,25 No such event is observed for the 1:1 mixture indicating that Na2(B12H12)0.5(B10H10)0.5 does not undergo a phase transition within this temperature window. Besides the first heating cycle, we also show DSC data of the third heating cycle, which almost coincides with the first cycle, and demonstrates the stability of the equimolar mixture when subjected to thermal cycling. Thermal characterization was also carried out for intermediate Na2(B12H12)1−x(B10H10)x compositions (0.1 < x < 0.6, see Fig. S1, ESI†) for which the endothermic peaks are still visible but decrease in size and ultimately vanish as the ratio approaches x = 0.5, at which the precursors fully react.
:1 equimolar mixture. Both precursors show a characteristic endothermic peak at the temperature of their respective structural phase transition, as previously described.24,25 No such event is observed for the 1:1 mixture indicating that Na2(B12H12)0.5(B10H10)0.5 does not undergo a phase transition within this temperature window. Besides the first heating cycle, we also show DSC data of the third heating cycle, which almost coincides with the first cycle, and demonstrates the stability of the equimolar mixture when subjected to thermal cycling. Thermal characterization was also carried out for intermediate Na2(B12H12)1−x(B10H10)x compositions (0.1 < x < 0.6, see Fig. S1, ESI†) for which the endothermic peaks are still visible but decrease in size and ultimately vanish as the ratio approaches x = 0.5, at which the precursors fully react.
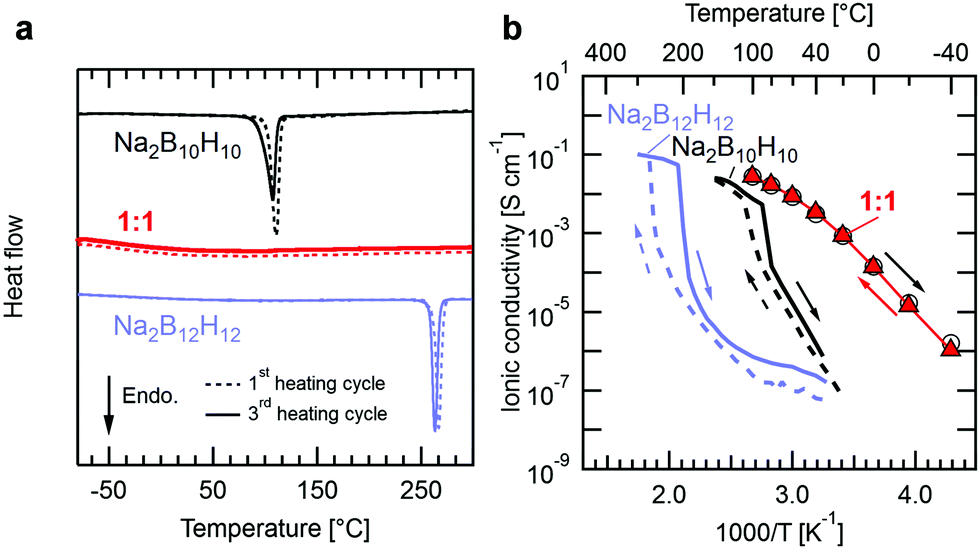 | ||
| Fig. 1 (a) DSC data for pure Na2B12H12, Na2B10H10 and their equimolar 1:1 mixture, Na2(B12H12)0.5(B10H10)0.5. (b) Temperature-dependent Na+ conductivity for pure Na2B12H12, Na2B10H1016,17 and Na2(B12H12)0.5(B10H10)0.5. Empty circles and filled triangles shown for the 1:1 mixture are measurement points upon heating and cooling, respectively. | ||
Fig. 1b presents the ionic conductivity of Na2(B12H12)0.5(B10H10)0.5 determined by impedance spectroscopy as a function of temperature (see Experimental section and Fig. S2, ESI,† for details). The conductivities of Na2B12H12 and Na2B10H10 obtained by Udovic et al.16,17 are also reported for comparison. The room temperature ionic conductivity for the equimolar ratio is several orders of magnitude higher than that of Na2B12H12 and Na2B10H10, reaching 0.9 mS cm−1 at 20 °C. This value is comparable to typical conductivities reported for oxide and sulfide electrolytes3–8 and only slightly lower than that of a standard sodium liquid electrolyte based on 1 M NaClO4 dissolved in EC:PC (3.3 mS cm−1 at 25 °C,26 assuming a sodium transference number of 0.4). Within the family of complex hydrides, this conductivity is comparable to the recently reported Na3BH4B12H12 and highly conducting, ball-milled Na2B12H1221,22 and is only surpassed by equimolar mixed carborate phases that can reach exceptional conductivity of 70 mS cm−1 at room temperature23 but have yet to demonstrate their applicability as SSEs in all-solid-state batteries. Similarly to the observed thermal behavior, the equimolar mixture shows no signs of a phase transition in the temperature-dependent conductivity data, whilst the conductivity of the precursors drops sharply at the onset temperatures at which latent heat is observed in DSC. Consequently, while hysteresis in conductivity is observed for both precursors upon temperature cycling, the conductivity of the equimolar mixture shows no hysteresis between heating and cooling. Still, the activation energy for Na+ diffusion in Na2(B12H12)0.5(B10H10)0.5 decreases smoothly from 0.7 at −40 °C to 0.4 eV at 100 °C with increasing temperature. A similar behavior was observed for carborate mixtures23 and Na3BH4B12H1221 and attributed to temperature dependent anion dynamics within the respective crystal structures. The connection between lattice parameter, anion dynamics and the evolution of cation conductivity in closo-borates was also recently discussed by Varley et al. based on ab initio molecular dynamics calculations.27
Crystal structure and lattice parameters were determined by X-ray diffraction (XRD). In Fig. 2a we compare the diffractograms of the equimolar mixture at room temperature with the ones of the precursors in their respective high temperature (HT) conductive phases, i.e. body-centered cubic (bcc) Na2B12H12 at 300 °C and face-centered cubic (fcc) Na2B10H10 at 180 °C. At room temperature the precursors show additional reflections indicative of the lower structure symmetry below the phase transition (see Fig. S3a, ESI†). We find that the reflections in the XRD pattern of the mixed phase can all be matched to the one of Na2B10H10 in its HT fcc phase. We conclude that the equimolar mixture stabilizes a single phase, isostructural to the highly conducting HT phase of Na2B10H10. Refinement yields a slightly larger unit cell parameter of a = 10.029 Å for Na2(B12H12)0.5(B10H10)0.5 at room temperature vs. a = 9.854 Å for Na2B10H10 at 180 °C (see Fig. S3b and c, ESI†). Additionally, XRD measurements of Na2(B12H12)0.5(B10H10)0.5 at 300 °C (see Fig. S3a, ESI†) show no structural change other than an expansion of the unit cell parameters, demonstrating again the high thermal stability in agreement with DSC and conductivity measurements. Note that equimolar mixtures of Na2B12H12 and Na2B10H10 were recently reported by Tang et al.,22 who also used mechanical milling followed by a heat treatment at 275 °C for 16 h. However, their mixture still contained predominantly (>70%) the low temperature (LT) phase of Na2B12H12 as refined from XRD, indicating only partial conversion to the mixed phase, and no conductivity measurement was reported. Our results prove that using appropriate reaction conditions, equimolar mixtures of Na2B12H12 and Na2B10H10 fully react and stabilize a single mixed-anion phase.
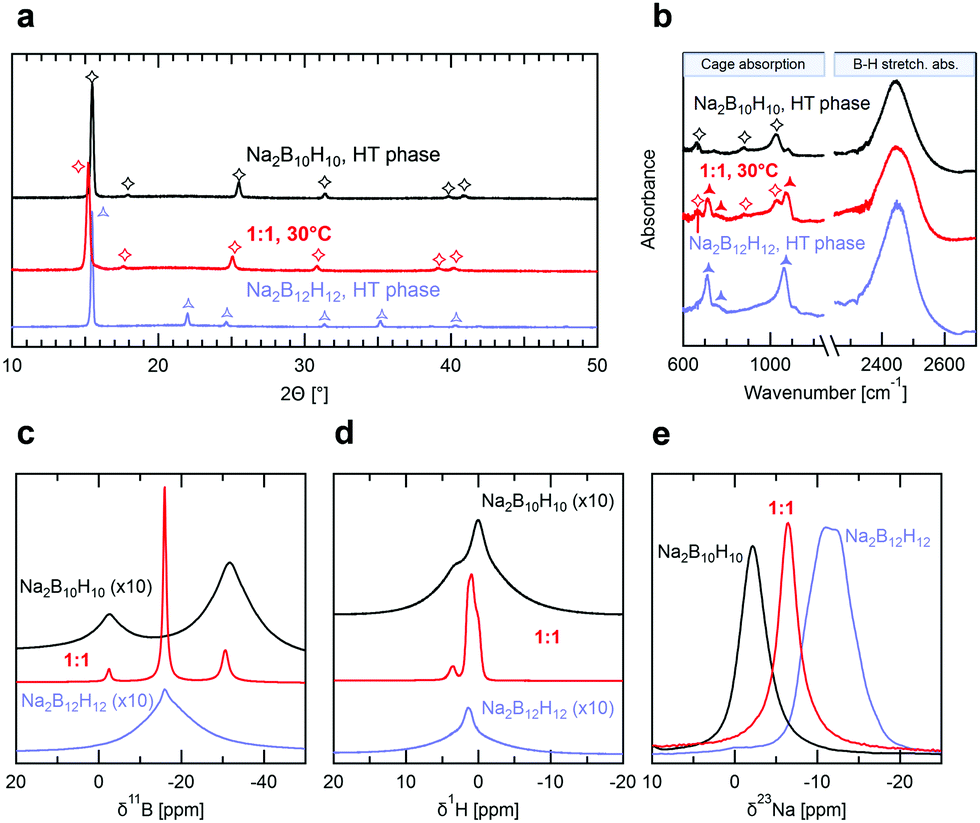 | ||
| Fig. 2 (a) XRD patterns and (b) infrared spectra of the HT phases of Na2B12H12 and Na2B10H10 recorded at 300 and 180 °C, respectively, compared to the room temperature phase of Na2(B12H12)0.5(B10H10)0.5. Characteristic features are marked by symbols. (c–e) Room temperature 1H, 11B and 23Na MAS solid-state NMR spectra of Na2B12H12, Na2B10H10 and Na2(B12H12)0.5(B10H10)0.5. For the 1H and 11B NMR data, the Na2B12H12 and Na2B10H10 spectra were Y-offset and up-scaled by a factor of 10 to facilitate data comparison. | ||
To gain insight into the local anion configuration in the mixed-anion structure, we performed infrared (IR) spectroscopy measurements. Fig. 2b compares the IR spectra of the HT phases of both Na2B12H12 (300 °C) and Na2B10H10 (180 °C) with Na2(B12H12)0.5(B10H10)0.5 at room temperature. For both precursors, B–H stretching and cage absorption modes are identified in the wavenumber ranges of 2400–2500 cm−1 and 600–1200 cm−1, respectively. These characteristic absorption modes correspond to those previously assigned in the IR spectra of B10H102− and B12H122− measured in solution,13,28 characteristic of the respective D4d and Ih point group symmetry of these ions. Recent theoretical DFT calculations confirm these assignments.29 In the LT phases of Na2B12H12 and Na2B10H10 (see Fig. S4, ESI†) significant splitting of the absorption modes can be seen, indicative of a lowered symmetry of the ions, whereas the anions in the HT phases adopt a similar symmetry than in the solution. Therefore, we conclude that they sit in a very symmetric environment that is a consequence of the cubic symmetry of the crystal structure and of the absence of a preferred orientation of the anions, i.e. of rotational disorder. The IR spectrum of Na2(B12H12)0.5(B10H10)0.5 at room temperature corresponds closely to the sum of the spectra of Na2B12H12 and Na2B10H10 in their respective HT phases and all vibration modes can be assigned to the two closo-borate anions. This shows that both B10H102− and B12H122− are still present in the structure and suggests either an average of rotationally disordered but static configurations, or of dynamic rotation of the anions.
To distinguish between these two cases, we recorded solid-state 11B, 1H and 23Na magic-angle spinning nuclear magnetic resonance (MAS NMR) spectra of Na2B12H12, Na2B10H10 and their 1:1 mixture. All samples were measured at room temperature to compare the ion dynamics. As seen from comparison in Fig. 2c–e, the spectrum of the equimolar mixture comprises all the resonances of the constituents in the precursors. Importantly, the resonances of the equimolar mixture are all considerably narrowed compared to the signals of the pure precursors, especially in the case of boron and hydrogen (Fig. 2c–e). This narrowing is a sign of fast dynamics of the ions in the 1:1 mixture compared to the borate starting materials. We may thus also interpret the IR data as a result of fast anion dynamics rather than an average of frozen configurations. Additionally, since X-ray diffraction already showed that Na2(B12H12)0.5(B10H10)0.5 has a well-defined crystal structure, we can conclude that this motion is rotational and not translational (see Fig. 3), as previously suggested based on NMR and quasi-elastic neutron scattering experiments on other closo-borates.30,3111B NMR also confirms that the B12H122− and B10H102− anions remain intact in Na2(B12H12)0.5(B10H10)0.5 with no significant 11B signal intensity detectable other than the ones of the closo-borate anions. The 1:1 B12H122− to B10H102− ratio was verified by 11B solution-state NMR by dissolving Na2(B12H12)0.5(B10H10)0.5 in deuterated water, and it was confirmed that both anions are indeed present in a 1:1 molar ratio (see Fig. S5, ESI†). Finally, it can be seen in Fig. 2e that the 23Na NMR chemical shift and thus the sodium environment in Na2(B12H12)0.5(B10H10)0.5 is averaged between that of Na2B10H10 and Na2B12H12, supporting the hypothesis of a solid solution phase, as depicted in Fig. 3.
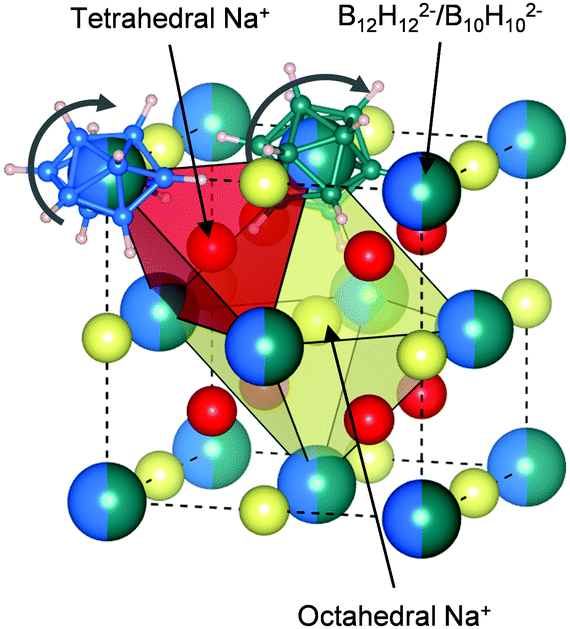 | ||
| Fig. 3 Simplified structure of Na2(B12H12)0.5(B10H10)0.5 based on the HT phase of Na2B10H10. Partially occupied Na+ ions sites are shown in different colors to distinguish tetrahedral and octahedral coordination. B12H122− and B10H102− anions are randomly distributed in the fcc framework. Two example anions are shown. | ||
To evaluate the suitability of Na2(B12H12)0.5(B10H10)0.5 as SSE in an all-solid-state sodium-ion battery, we performed cyclic voltammetry tests at room temperature. Fig. 4a shows current density vs. voltage measured for a Na|Na2(B12H12)0.5(B10H10)0.5|Pt cell cycled at voltages between −0.5 V and 6.5 V at 0.1 mV s−1. Near 0 V, we observed characteristic anodic and cathodic current peaks associated with sodium stripping and plating. Towards higher voltages, the current density remains low up to 6.5 V but first oxidation currents were observed in the nA cm−2 range. We therefore built two additional cells for a precise measure of the electrochemical stability window of Na2(B12H12)0.5(B10H10)0.5. To determine its oxidative stability, a platinum working electrode (WE) was swept against a sodium counter electrode from the open-circuit voltage of 1.6 V up to a voltage of 5.5 V at a slow rate of 0.1 mV s−1 to remain near equilibrium. Results are shown in Fig. 4b. Small currents indicative of slow electrolyte oxidation can be measured for cell voltages above 3 V but remain below 10 nA cm−2 at higher voltages. To determine the reductive stability, an aluminum working electrode, which unlike platinum does not alloy with sodium, was preferred. Starting at the cell's open circuit voltage of 1.6 V, the voltage was swept down to −0.5 V. On aluminum, no reduction currents are observed down to 0 V, below which the sodium plating peak appears, suggesting that the electrolyte is stable in contact with metallic sodium.
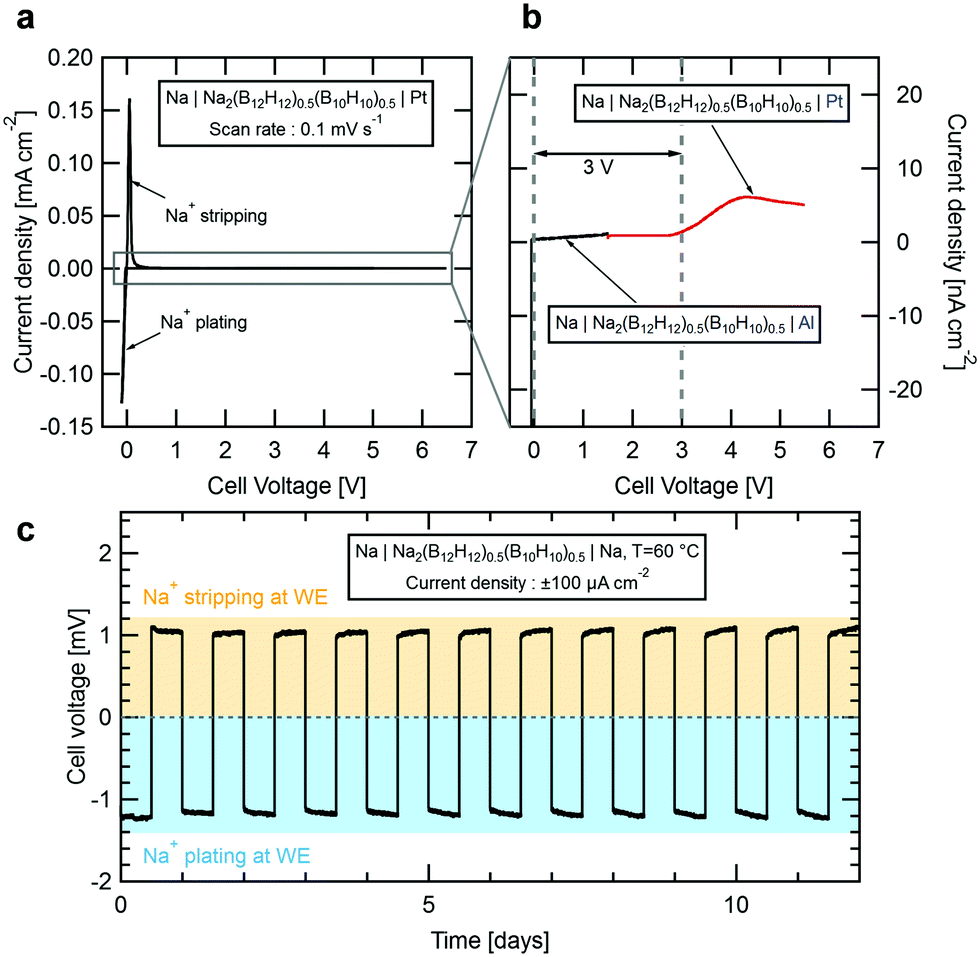 | ||
| Fig. 4 Room temperature cyclic voltammogram of (a) a Na|Na2(B12H12)0.5(B10H10)0.5|Pt cell from −0.5 to 6.5 V at a scan rate of 0.1 mV s−1 and (b) of Na|Na2(B12H12)0.5(B10H10)0.5|Al & Na|Na2(B12H12)0.5(B10H10)0.5|Pt cells between −0.5 and 5.5 V showing the oxidative (red) and reductive (black) stability of the electrolyte, respectively. (c) Galvanostatic cycling of a symmetric Na|Na2(B12H12)0.5(B10H10)0.5|Na cell at 60 °C with a current density of 100 μA cm−2. | ||
We conclude that Na2(B12H12)0.5(B10H10)0.5 offers a practical electrochemical stability window of 3 V, most likely limited by oxidation of the B10H102− anions.32 Preliminary cyclic voltammetry measurements performed on other closo-borate electrolytes suggested a much larger stability window for this class of compounds.17,21,22 However, we believe that the stability has been over-estimated due to the very small oxidation currents that may not have been detected in previous measurements. In fact, Han et al.33 recently discussed the limitation of conventional cyclic voltammetry measurements to evaluate the true stability window of solid-state electrolytes. This limitation can be overcome only if one is able to measure very small currents (we used a low current channel with pA resolution) or by artificially increasing the contact area between the electrolyte and the working electrode.
To further confirm the compatibility with a sodium metal anode, we built a symmetric Na|Na2(B12H12)0.5(B10H10)0.5|Na cell which was galvanostatically cycled at 60 °C. Fig. 4c shows the evolution of the cell voltage over time as sodium is plated and stripped at a current of ±100 μA cm−2 in intervals of 12 h. Our results demonstrate that sodium is plated and stripped fully reversibly with a low combined over-potential reaching a maximum of 1.2 mV, in good agreement with the measured conductivity for the electrolyte at 60 °C. Importantly, the overall voltage remains stable for 12 days confirming that the electrolyte is not degrading. Note that during a 12 h cycle, a charge equivalent of 1.2 mA h cm−2 is cycled through the material. Considering a typical sodium cathode material with a capacity of 120 mA h g−1 and a mass loading of 20 mg, each 12 h cycle of this measurement corresponds to 50% of a full charge.
In conclusion, we demonstrated that among SSEs with comparable conductivity, Na2(B12H12)0.5(B10H10)0.5 stands out as a particularly stable material, notably when compared to sulphide phases. The electrochemical stability window of 3 V is compatible with high performance sodium cathode materials such as Na2FePO4F, NaCrO2. We further demonstrated that Na2(B12H12)0.5(B10H10)0.5 is compatible with a sodium metal anode enabling long-term and reversible sodium plating and stripping. We also believe that further anion engineering e.g. by mixing larger halogenated closo-borates with a lower charge density, could yield even higher stability and conductivity and that this class of materials holds promise for competitive high voltage all-solid-state sodium-ion batteries.
We would like to thank the Swiss National Science Foundation for financial support within the Sinergia project ‘Novel ionic conductors’ under contract number CRSII2_160749/1. The NMR hardware was partially granted by the Swiss National Science Foundation (SNFS, grant no. 206021_150638/1).
Notes and references
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3432 CrossRef PubMed.
- H. Engstrom, J. B. Bates, W. E. Brundage and J. C. Wang, Solid State Ionics, 1981, 2, 265 CrossRef CAS.
- J. B. Goodenough, H. Y.-P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203 CrossRef CAS.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- L. Zhang, K. Yang, J. Mi, L. Lu, L. Zhao, L. Wang, Y. Li and H. Zeng, Adv. Energy Mater., 2015, 5, 2 Search PubMed.
- A. Banerjee, K. H. Park, J. W. Heo, Y. J. Nam, C. K. Moon, S. M. Oh, S.-T. Hong and Y. S. Jung, Angew. Chem., Int. Ed., 2016, 55, 9634 CrossRef CAS PubMed.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266 CrossRef CAS.
- W. D. Richards, T. Tsujimura, L. J. Miara, Y. Wang, J. C. Kim, S. P. Ong, I. Uechi, N. Suzuki and G. Ceder, Nat. Commun., 2016, 7, 11009 CrossRef CAS PubMed.
- I.-H. Chu, C. S. Kompella, H. Nguyen, Z. Zhu, S. Hy, Z. Deng, Y. S. Meng and S. P. Ong, Sci. Rep., 2016, 6, 33733 CrossRef CAS PubMed.
- R. Mohtadi, A. Remhof and P. Jena, J. Phys.: Condens. Matter, 2016, 28, 353001 CrossRef PubMed.
- B. R. S. Hansen, M. Paskevicius, H.-W. Li, E. Akiba and T. R. Jensen, Coord. Chem. Rev., 2016, 323, 60 CrossRef CAS.
- E. L. Muetterties, J. H. Balthis, Y. T. Chia, W. H. Knoth and H. C. Miller, Inorg. Chem., 1964, 3, 444 CrossRef CAS.
- S. Giri, S. Behera and P. Jena, Angew. Chem., Int. Ed., 2014, 53, 13916 CrossRef CAS PubMed.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900 CrossRef CAS PubMed.
- T. J. Udovic, M. Matsuo, A. Unemoto, N. Verdal, V. Stavila, A. V. Skripov, J. J. Rush, H. Takamura and S. Orimo, Chem. Commun., 2014, 50, 3750 RSC.
- T. J. Udovic, M. Matsuo, W. S. Tang, H. Wu, V. Stavila, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, J. J. Rush, A. Unemoto, H. Takamura and S. Orimo, Adv. Mater., 2014, 26, 7622 CrossRef CAS PubMed.
- W. S. Tang, A. Unemoto, W. Zhou, V. Stavila, M. Matsuo, H. Wu, S. Orimo and T. J. Udovic, Energy Environ. Sci., 2015, 8, 3637 CAS.
- W. S. Tang, M. Matsuo, H. Wu, V. Stavila, W. Zhou, A. A. Talin, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, A. Unemoto, S.-I. Orimo and T. J. Udovic, Adv. Energy Mater., 2016, 6, 1502237 CrossRef.
- L. He, H.-W. Li, H. Nakajima, N. Tumanov, Y. Filinchuk, S.-J. Hwang, M. Sharma, H. Hagemann and E. Akiba, Chem. Mater., 2015, 27, 5483 CrossRef CAS.
- Y. Sadikin, M. Brighi, P. Schouwink and R. Černý, Adv. Energy Mater., 2015, 5, 1501016 CrossRef.
- W. S. Tang, M. Matsuo, H. Wu, V. Stavila, A. Unemoto, S. Orimo and T. J. Udovic, Energy Storage Mater., 2016, 4, 79 CrossRef.
- W. S. Tang, K. Yoshida, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, M. Dimitrievska, V. Stavila, S. Orimo and T. J. Udovic, ACS Energy Lett., 2016, 1, 659 CrossRef CAS.
- B. Bonnetot, H. Mongeot, A. Aboukhassib and F. Lefebvre, Inorg. Chim. Acta, 1992, 193, 21 CrossRef CAS.
- N. Verdal, J.-H. Her, V. Stavila, A. V. Soloninin, O. A. Babanova, A. V. Skripov, T. J. Udovic and J. J. Rush, J. Solid State Chem., 2014, 212, 81 CrossRef CAS.
- A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. R. Palacín, Energy Environ. Sci., 2012, 5, 8572 CAS.
- J. B. Varley, K. Kweon, P. Mehta, P. Shea, T. W. Heo, T. J. Udovic, V. Stavila and B. C. Wood, ACS Energy Lett., 2017, 2, 250 CrossRef CAS.
- E. L. Muetterties, R. E. Merrifield, H. C. Miller, W. H. Knoth and J. R. Downing, J. Am. Chem. Soc., 1962, 84, 2506 CrossRef CAS.
- D. Sethio, L. M. Lawson Daku and H. Hagemann, Int. J. Hydrogen Energy, 2016, 41, 6814 CrossRef CAS.
- N. Verdal, T. J. Udovic, V. Stavila, W. S. Tang, J. J. Rush and A. V. Skripov, J. Phys. Chem. C, 2014, 118, 17483 CAS.
- A. V. Soloninin, M. Dimitrievska, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, W. S. Tang, V. Stavila, S. Orimo and T. J. Udovic, J. Phys. Chem. C, 2017, 121, 1000 CAS.
- J. H. Morris, H. J. Gysling and D. Reed, Chem. Rev., 1985, 85, 51 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: 10.1039/c7cc00794a |
| This journal is © The Royal Society of Chemistry 2017 |