
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rigidly linking cyclometallated Ir(III) and Pt(II) centres: an efficient approach to strongly absorbing and highly phosphorescent red emitters†
Graeme
Turnbull
 a,
J. A. Gareth
Williams
*b and
Valery N.
Kozhevnikov
*a
a,
J. A. Gareth
Williams
*b and
Valery N.
Kozhevnikov
*a
aDepartment of Applied Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: valery.kozhevnikov@northumbria.ac.uk
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 16th February 2017
Abstract
The synthesis and photophysical properties of an unprecedented tetranuclear complex are described, in which a fac-tris-cyclometallated Ir(III) centre is rigidly connected to three cyclometallated Pt(II) centres. The complex absorbs strongly up to ∼600 nm and emits red light with unusually high efficiency.
Phosphorescent metal complexes that emit light with high efficiency are being explored for a variety of applications. For example, some are used as phosphors in commercial organic light-emitting diodes (OLEDs), whilst ionic variants are under investigation for light-emitting electrochemical cells (LEECs).1,2 Meanwhile, their relatively long luminescence lifetimes and good photostability have rendered them of interest as probes for bio-imaging,3 particularly for time-resolved detection procedures,4 and for sensing of biologically and environmentally relevant analytes such as metal ions, molecular oxygen and organic vapours.5,6 The most successful complexes have tended to be based around cyclometallated iridium complexes, where the high spin–orbit coupling associated with a 3rd-row metal ion in a pseudo-octahedral geometry typically ensures that the formally forbidden T1 → S0 phosphorescence is promoted effectively.7 The brightest such emitters emit in the green region of the spectrum; e.g., the archetypal complex fac-Ir(ppy)3 emits in degassed solution8 with a λmax of 508 nm and a photo-luminescence quantum yield recently re-assessed to be 0.97.‡
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8c
8c
Strategies for shifting the emission of this family of compounds to the red and near-infrared rely on increasing the conjugation within the ligands, and/or on the use of more electron-rich cyclometallating rings (e.g. thienyl in place of phenyl).9,10 Whilst such methods do lead to desired red shifts, efficiencies invariably fall off compared to green emitters, owing to the combined effects of increased non-radiative decay of lower-energy excited states, and the decrease in radiative rate constants. The latter effect can be understood not only in terms of the ν3 term within the Einstein coefficient for spontaneous emission (which affects all emitters) but also through the fact that the extent of participation of the metal in the excited state necessarily decreases as the ligands become more electron rich and/or the conjugation increases. The effect may even be manifest through such complexes displaying fluorescence from the singlet excited state, in competition with triplet state formation and subsequent phosphorescence.10
We have recently shown how the introduction of a second iridium centre to generate dinuclear complexes can lead to efficient red emitters that display unusually high triplet radiative decay rates, kr, and hence high phosphorescence quantum yields.11 The complexes feature two iridium ions each bound to a common, bridging heterocyclic ring, such as a pyrimidine, within a bi- or tridentate ligand. Similar results were found for related dinuclear platinum complexes, whose emission was red-shifted and kr values increased relative to mononuclear analogues.12
Given the importance of fac-Ir(ppy)3 in the field, we have now sought to modify this archetypal complex in such a way that a Pt(II) ion is introduced into each ligand, to generate an unprecedented heterometallic tetranuclear complex, 1 (Fig. 1).
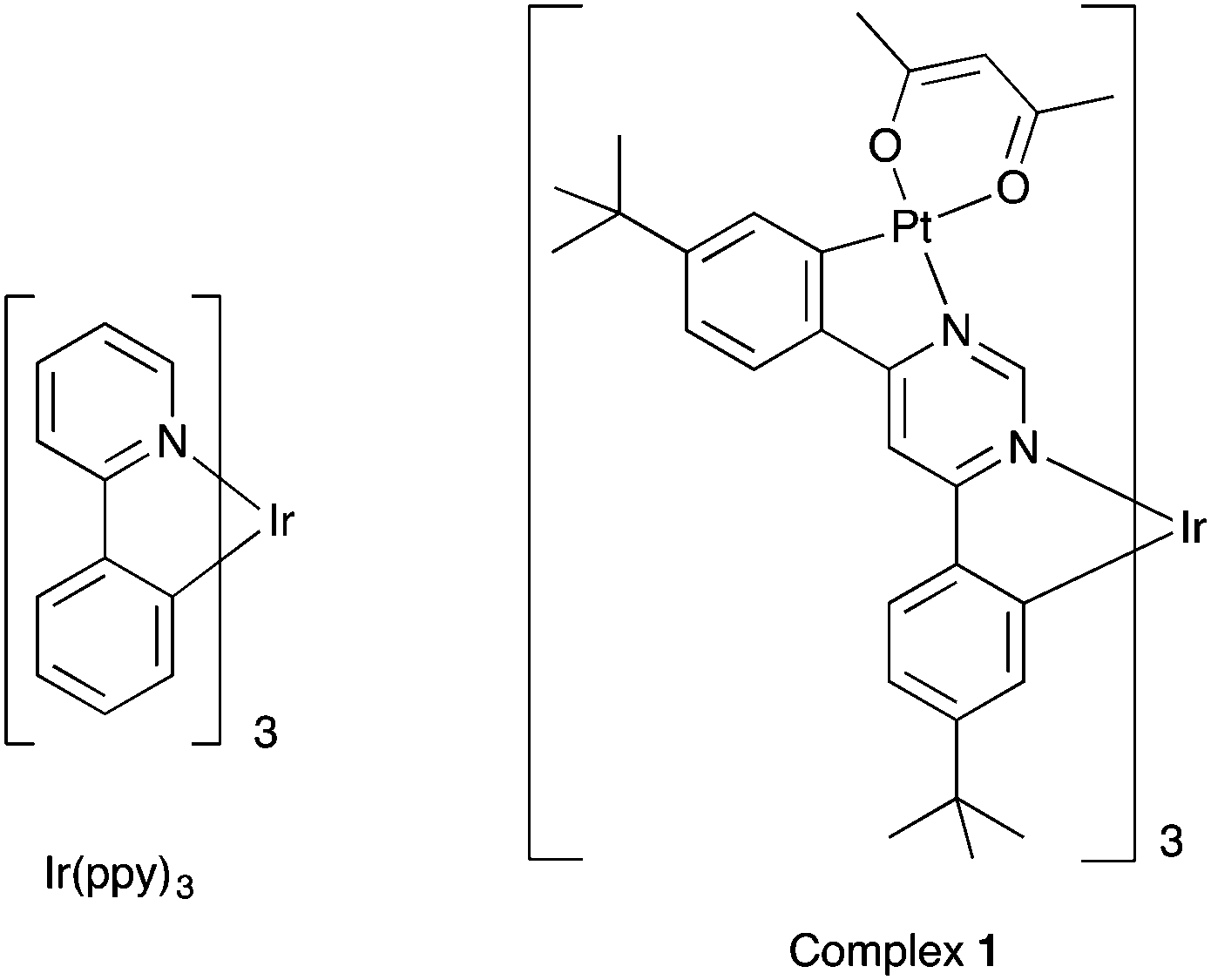 | ||
| Fig. 1 Structures of Ir(ppy)3 and of the heterometallic tetranuclear complex 1. | ||
Our strategy relies on the use of 4,6-bis-(4-t-butylphenyl)pyrimidine (bppymH2) as a bis-N^C-coordinating ligand that can bind simultaneously to two metal ions through cyclometallation.12atert-Butyl substituents were included in the para positions of the phenyl rings, in order to improve the solubility of the final complexes.
The target complex Ir{(μ-bppym)Pt(acac)}31 was obtained in two steps (Scheme 1). Owing to the harsh conditions required for the preparation of fac tris-cyclometallated Ir(III) complexes, we chose to incorporate the iridium centre first, to be followed by the introduction of the platinum ions. The fac tris-cyclometallated complex Ir(bppymH)32 was initially obtained in 30% yield from the corresponding bis-cyclometallated complex Ir(bppymH)2(acac) 3 upon reaction with bppymH2 in glycerol at 200 °C, ESI.† Compound 3 is itself prepared through the intermediacy of [Ir(bppymH)2(μ-Cl)]2, obtained upon reaction of bppymH2 with IrCl3·3H2O in a 2:1 molar ratio, followed by treatment with sodium acetylacetonate. An alternative, more direct and higher-yielding synthesis of 2 proved to be the reaction of bppymH2 with Ir(acac)3 in the presence of o-phosphoric acid, which gave the product in 52% yield after column chromatography.
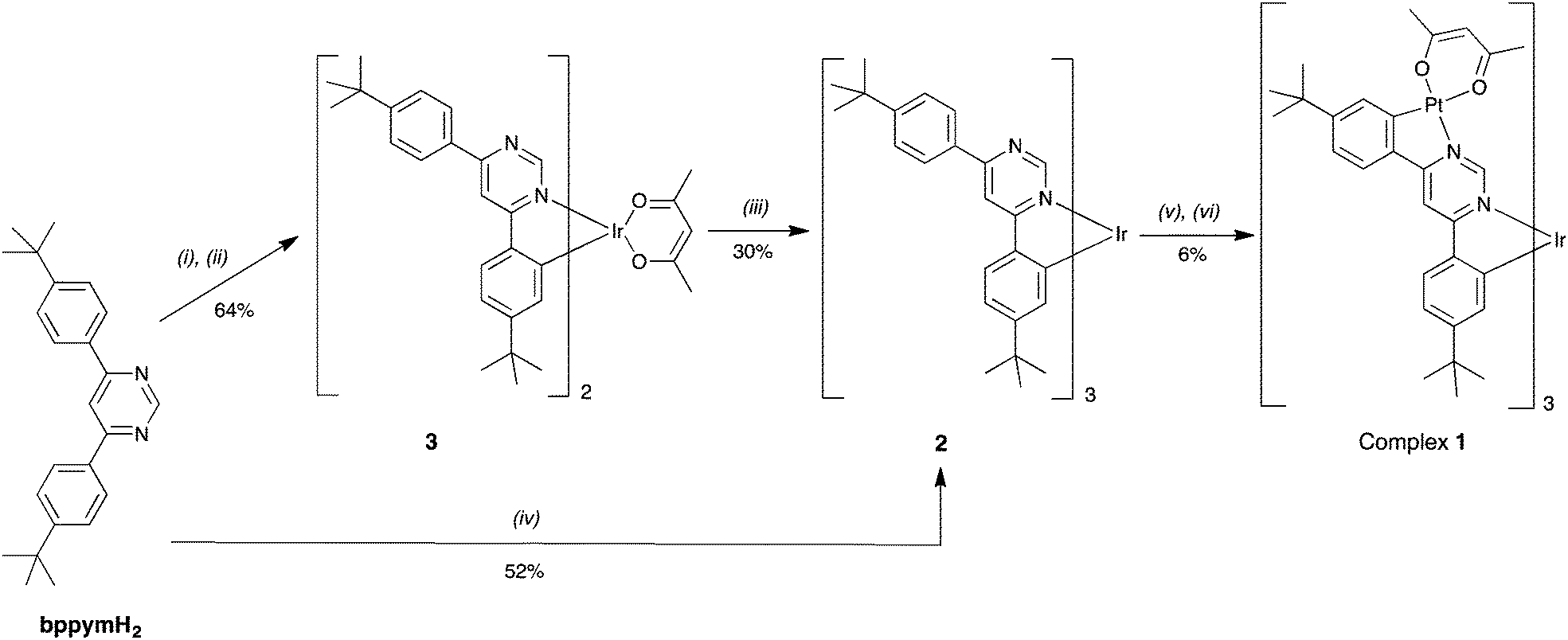 | ||
| Scheme 1 Routes to 1 starting from bppymH2: (i) IrCl3·3H2O (0.5 equiv.), ethoxyethanol, 110 °C, 14 h. (ii) Na(acac), 90 °C, 5 h. (iii) bppymH2, glycerol, 200 °C, 2 h. (iv) Ir(acac)3 (0.28 equiv.), H3PO4, glycerol, 190 °C, 18 h. (v) K2PtCl4 (>3 equiv.), AcOH/MeCN, 36 h. (vi) Na(acac), acetone, reflux, 10 h. | ||
The mononuclear complex 2 was then reacted with K2PtCl4 (3.75 equiv.) in a mixture of acetic acid and acetonitrile at reflux, followed by treatment of the resulting solid with an excess of sodium acetylacetonate in acetone, leading to the target tetranuclear complex 1 in 6% yield. The low yield may be related to the need for a total of three such metallations and subsequent chloride metatheses to occur.§ The fac geometry around the Ir centre ensures that the three Pt units are able to be accommodated on one side of the molecule without excessive steric hindrance, despite the large size and apparently congested nature of the molecule. Molecular mechanics and DFT calculations confirm that the structure is feasible, with the distances between neighbouring ligands being no shorter than van der Waals contacts (see ESI,† including Fig. S2.1 for 3D views of the molecule).
The UV-visible absorption spectrum of the new tetranuclear complex 1 in dichloromethane solution at room temperature is shown in Fig. 2, together with the spectra of the mononuclear complex 2 as a model and of fac-Ir(ppy)3. Corresponding data are provided in Table 1. It can be seen that the absorption of complex 1 is strongly red-shifted compared to fac-Ir(ppy)3 – by around 4500 cm−1 for most of the bands – and that the extinction coefficients are greatly increased, by a factor of around 4, throughout the spectrum. By reference to the model complex 2, it is evident that one contributing factor in generating these effects is the change from a pyridine to a pyrimidine ring in the N^C-cyclometallating ligand. This is readily rationalised since the lowest-energy transitions in Ir(N^C)3 complexes are normally associated with charge-transfer transitions in which the heterocyclic ring is the acceptor.1c,9,13 However, the introduction of the Pt(II) centres onto the second N^C unit of each ligand leads to a further significant red shift of the bands in the visible region – and indeed also in the UV – of around 2000 cm−1. Moreover, the molar absorptivity is further increased; for example, ε for the most intense band in the visible region of each complex is increased by a factor of 2 (for complex 1, λmax = 455 nm, ε = 49600 M−1 cm−1; for complex 2, λmax = 416 nm, ε = 27500 M−1 cm−1). It may be noted that there are no bands with ε > 10000 M−1 cm−1 in the visible region for fac-Ir(ppy)3.
 | ||
| Fig. 2 Absorption spectra of 1 (red solid line), 2 (blue) and fac-Ir(ppy)3 (green) in CH2Cl2 at 298 ± 3 K. The excitation spectrum of 1 under the same conditions, registered at λem = 610 nm, is the red dotted line labelled 1ex. | ||
| Complex | Absorptiona | Emission | Φ lum , | τ /ns | k r /105 s−1 | ∑knre/105 s−1 |
f/108 M−1 s−1 |
Emission at 77 Kg | |
|---|---|---|---|---|---|---|---|---|---|
| λ max/nm (ε/103 M−1 cm−1) | λ max/nm | λ max/nm | τ/ns | ||||||
|
a Bands of λ > 250 nm are listed.
b In degassed solution.
c Luminescence quantum yield measured for 1 and 2 using [Ru(bpy)3]Cl2 as the standard.
d In degassed solution for 1 and 2, under nitrogen for Ir(ppy)3; values in air-equilibrated solution are shown in parenthesis.
e
k
r and ∑knr are the radiative and non-radiative rate constants estimated according to: kr = Φlum/τ and knr = (1 − Φlum)/τ.
f Bimolecular rate constant for quenching by molecular oxygen, estimated from the τ values in degassed and air-equilibrated solution, and assuming [O2] = 2.2 mM in CH2Cl2 at atmospheric pressure of air.
g Data for 1 and 2 at 77 K are in a glass of diethyl ether/isopentane/ethanol, 2:2:1 v/v.
h Data refer to the fac isomer of Ir(ppy)3.
i Absorption data for Ir(ppy)3 in CH2Cl2, from ref. 8b.
j Emission data for Ir(ppy)3 are in 2-MeTHF except for τ in air-equilibrated solution, which is in toluene; no kQ value is therefore given; data from ref. 8c. Φlum was reported in ref. 8a and b to be 0.40.
|
|||||||||
| 1 | 255 (97.7), 334 (121), 404 (41.6), 455 (49.6), 554 (12.5) | 611 | 0.76 | 720 [300] | 11 | 3.3 | 8.8 | 586, 635 | 2600 |
| 2 | 260sh (66.0), 279 (73.5), 313 (102), 416 (27.5), 506 (9.15) | 590 | 0.61 | 1400 [200] | 4.4 | 2.8 | 19 | 556, 593 | 3900 |
| Ir(ppy)3 , , | 283 (44.8), 341 (9.2), 377 (12.0), 405 (8.1), 455 (2.8), 488 (1.6) | 508 | 0.97 | 1600 [36] | 6.1 | 0.2 | — | 491 | 4000 |

TD-DFT calculations confirm the observed red-shift in the lowest-energy absorption bands in 1 compared to 2 (see ESI†), and suggest that the effect upon introduction of the platinum centres is due to a stabilisation of the LUMO that exceeds that of the HOMO. Thus, the LUMO energies of 1 and 2 are calculated to be −1.90 and −2.27 eV respectively (a stabilisation of 0.37 eV), whilst the HOMO values are −5.12 and −5.34 V (stabilisation of 0.22 eV). A large stabilisation of the LUMO upon platination is confirmed by cyclic voltammetry (see Fig. S3.1 of the ESI†). Complex 1 shows a first reduction at −2.22 V (in CH2Cl2versus ferrocene) whereas 2, in common with complexes like Ir(ppy)3, shows no reduction within the accessible window (the region down to −3 V in CH2Cl2 was probed). The first oxidation, in contrast, is shifted by <0.3 V.
Complex 1 is intensely luminescent in solution at room temperature, emitting in the red region of the spectrum; λmax = 611 nm (Fig. 3 and Table 1). The luminescence quantum yield of 0.76 renders it one of the very brightest red-emitting molecular phosphors reported.9 The emission is strongly red-shifted compared to fac-Ir(ppy)3 by over 3000 cm−1, though the shift in λmax relative to 2 is only around 500 cm−1 at room temperature (around 1000 cm−1 at 77 K). Inspection of the room-temperature spectra reveals that the polynuclear complex displays a narrower profile than that of the mononuclear model under these conditions (half-height widths of 2170 and 2860 cm−1 respectively). Meanwhile, the Stokes shift between the lowest-energy absorption band and the peak of the emission band is significantly smaller for the polynuclear system (around 1600 cm−1 for 1 compared to 2800 cm−1 for 2). Both observations are suggestive of the polynuclear system being more rigid and subject to less excited-state distortion. It is notable that, at 77 K, the ratio of the intensity of the 0,0 to 0,1 vibrational components is higher for 1 than for 2, an observation that also suggests less excited-state distortion for 1.14 The luminescence lifetime τ of complex 1 is 720 ns at room temperature, an unusually short lifetime for cyclometallated iridium complexes (e.g., for fac-Ir(ppy)3, τ = 1.9 μs under comparable conditions) yet the luminescence quantum yield remains high. The short lifetime is thus indicative of an unusually fast radiative decay rate kr (as opposed to deleterious non-radiative decay). Assuming that the emitting state is formed with unitary efficiency upon light absorption, kr can be estimated from τ and Φlum to be 11 × 105 s−1, which is double that of fac-Ir(ppy)3. The substantial augmentation of kr is consistent with the effect of additional metal ions in polynuclear systems we have investigated previously.11,12 Whilst it may be tempting to attribute the effect to enhanced spin–orbit coupling associated with the presence of the additional metal ions, the fact that the spin-allowed bands in absorption are also significantly increased in intensity (higher ε values in the polynuclear system) suggests that there may be other factors at work in increasing oscillator strengths.
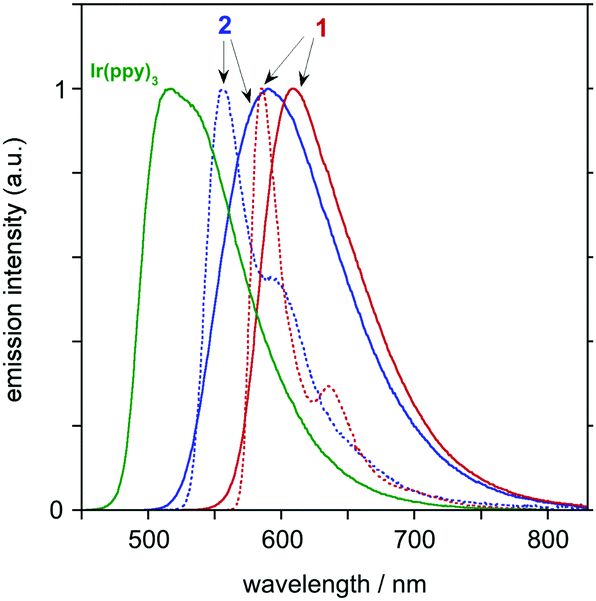 | ||
| Fig. 3 Emission spectra of 1, 2 and fac-Ir(ppy)3 in CH2Cl2 at 298 ± 3 K (red, blue and green solid lines respectively), together with the spectra of 1 and 2 at 77 K in diethyl ether/isopentane/ethanol (2:2:1 v/v). | ||
Complex 1 is much less sensitive to quenching by dissolved molecular oxygen than typical iridium-based emitters; e.g., τ and Φlum are decreased by a factor of only 2 upon aeration {which compares with 7-fold for 2 and around 13-fold for fac-Ir(ppy)3}. The lower sensitivity to quenching can be attributed not only to the shorter lifetime, but also to a smaller value of the bimolecular rate constant for quenching, kQ (Table 1). This in turn may be associated, at least in part, with the substantially larger molecular weight of the polynuclear complex.¶
In summary, the preparation of the new heterometallic tetranuclear complex demonstrates how the introduction of additional heavy metal ions can allow access to phosphorescent molecules that (i) emit with higher efficiency in the red region of the spectrum with narrower spectral profiles, and (ii) absorb out to longer wavelengths and with substantially higher extinction coefficients than conventional mononuclear systems. Point (i) arises from an increase in radiative rate constants associated with the additional metal ions, a trend that is emerging from homo-dinuclear systems too.11,12 However, in the present instance at least, it may also be related to an increase in the rigidity of the system, helping to minimise excited state distortion and hence reduce competitive non-radiative decay. High rigidity also leads to the narrow spectral profile, important for colour purity in display screen applications. These observations point a way for the development of more efficient red and NIR molecular emitters.
Meanwhile, point (ii) is potentially important with regards to the use of iridium complexes for solar energy conversion and photocatalysis, for example, where more efficient absorption across the visible region of the spectrum is desirable for practicable application.15 Finally, we note that the combination of efficient low-energy [red] absorption and low-energy [red/NIR] emission is a key desirable criterion for bioimaging applications, to exploit the “window of transparency” of biological tissue in this region of the spectrum.3,4
Complex 1 may be regarded as a metallodendrimer of first-generation type, where the cyclometallated Pt(II) shell surrounds the central fac-tris-cyclometallated Ir(III) core. Many other such multinuclear complexes can be envisaged that may offer attractive optical or opto-catalytic properties.
We thank Melissa Walden, Emma Puttock and Dr Marcus Durrant for their valued input, and the Universities of Northumbria and Durham for support.
Notes and references
- (a) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (b) Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC; (c) L. F. Gildea and J. A. G. Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013 Search PubMed; (d) E. Baranoff and B. F. E. Curchod, Dalton Trans., 2015, 44, 8318 RSC.
- (a) R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed; (b) S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985 CrossRef CAS.
- (a) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS; (b) K. K.-W. Lo and S. P.-Y. Li, RSC Adv., 2014, 4, 10560 RSC; (c) M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2014, 50, 384 RSC; (d) Z. Q. Guo, W.-L. Tong and M. C. W. Chan, Chem. Commun., 2014, 50, 1711 RSC.
- (a) E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879 RSC; (b) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bonding, 2015, 165, 205 CrossRef CAS.
- (a) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC; (b) Y. You, S. Cho and W. Nam, Inorg. Chem., 2014, 53, 1804 CrossRef CAS PubMed.
- (a) X.-D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666 RSC; (b) T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148 CrossRef CAS PubMed; (c) J. R. Berenguer, J. Fernandez, B. Gil, E. Lalinde and S. Sanchez, Chem. – Eur. J., 2014, 20, 2574 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS.
- (a) K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431 CrossRef CAS; (b) A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. M. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed; (c) T. Sajato, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard III and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813 CrossRef PubMed.
- (a) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed; (b) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshini and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed; (c) Y. J. Su, H. L. Huang, C. L. Li, C. H. Chien, Y. T. Tao, P. T. Chou, S. Datta and R. S. Liu, Adv. Mater., 2003, 15, 224 CrossRef; (d) F. M. Hwang, H. Y. Chen, P. S. Chen, C. S. Liu, Y. Chi, C. F. Shu, F. I. Wu, P. T. Chou, S. M. Peng and G. H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef CAS PubMed.
- (a) D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804 CrossRef CAS PubMed; (b) P.-T. Chou, Y. Chi, M.-W. Chung and C.-C. Lin, Coord. Chem. Rev., 2011, 255, 2653 CrossRef CAS.
- (a) P.-H. Lanoë, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831 RSC; (b) R. E. Daniels, S. Culham, M. Hunter, M. C. Durrant, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Dalton Trans., 2016, 45, 6949 RSC.
- (a) V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304 CrossRef CAS PubMed; (b) S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef CAS PubMed.
- P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634 CrossRef CAS.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed.
- For example: (a) D. N. Chirdon, W. J. Transue, H. N. Kagalwala, A. Kaur, A. B. Maurer, T. Pintauer and S. Bernhard, Inorg. Chem., 2014, 53, 1487 CrossRef CAS PubMed; (b) S. Muhmel, D. Alpers, F. Hoffmann and M. Brasholz, Chem. – Eur. J., 2015, 21, 12308 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic methods, compound characterisation, TD-DFT and electrochemical data. See DOI: 10.1039/c7cc00656j |
| ‡ In deoxygenated 2-MeTHF at 298 K. |
| § The synthesis and isolation of the corresponding trinuclear (Pt2Ir) and dinuclear (PtIr) complexes has been attempted, using ratios of K2PtCl4 to complex 2 of proportionately less than 3:1. Whilst 1H NMR and MS data did imply the formation of these species (and hence that the formation of 1 from 2 likely proceeds via mono- and bis-platinated intermediates), it has not proved possible to isolate pure samples, apparently due to their co-elution under a variety of chromatographic conditions. |
¶ According to the Smoluchowski equation,  is proportional to the diffusion coefficient, which is in turn proportional to √MW through Fick's Law. The MW values of 1 and 2 are 2100 and 1221 respectively. is proportional to the diffusion coefficient, which is in turn proportional to √MW through Fick's Law. The MW values of 1 and 2 are 2100 and 1221 respectively. |
| This journal is © The Royal Society of Chemistry 2017 |