
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A D3h-symmetry hexaazatriphenylene-tris-N-heterocyclic carbene ligand and its coordination to iridium and gold: preliminary catalytic studies†
S.
Ibáñez
,
M.
Poyatos
* and
E.
Peris
 *
*
Institute of Advanced Materials (INAM), Universitat Jaume I, Av. Vicente Sos Baynat s/n, Castellón 12071, Spain. E-mail: poyatosd@uji.es; eperis@uji.es
First published on 27th February 2017
Abstract
A new D3h-symmetry tris-N-heterocyclic carbene ligand has been prepared and coordinated to iridium and gold. The ligand contains an electron-poor hexaazatriphenylene core; thus, the resulting tris-NHC ligand is a poor electron donor. The tris–Au(I) complex was tested in the hydroamination of terminal alkynes and in the three-component Strecker reaction.
Hexaazatriphenylene (HAT) constitutes an attractive building block for supramolecular systems and for molecular coordination compounds because it combines exceptional topological and electronic features with great coordination ability. HAT is the smallest two-dimensional N-containing polycyclic aromatic system. It has been used as building block for a myriad of supramolecular systems with a variety of applications.1 Due to the presence of three geometrically independent chelating coordination sites, which are useful for the generation of self-assembled species, HAT has been extensively used for the preparation of a large number of molecular, supramolecular and polymer-based coordination species.2
During the last few years, we have been interested in designing poly-N-heterocyclic carbene ligands3 connected by polycyclic aromatic hydrocarbons, aiming to facilitate the preparation of homogeneous metal-based catalysts with enhanced properties. In the course of our research, we observed that the use of this type of ligands induced interesting catalytic properties that we mainly attributed to substrate–ligand supramolecular interactions,4 and they therefore constitute a subclass of the recently classified smart NHC ligands.5 Among the NHC-based ligands that we obtained, the triphenylene-tris-NHC ligand (A) displayed unique topological features because it allowed the coordination of three metal fragments in a pseudo-D3h symmetry environment,6 thus being the only tris-NHC ligand reported to date able to facilitate this type of geometry. The presence of the triphenylene core allowed the supramolecular interaction of the ligand with the substrates, affording catalysts that performed better than their related monometallic analogues lacking this polycyclic aromatic core.6a The same tris-NHC ligand was also used as scaffold for the preparation of main-chain organometallic polymers, therefore also showing potential to afford three-dimensional structures.7 Prompted by these precedents, we became interested in merging the properties associated with planar and rigid tris-NHC ligands with the presence of a HAT core. We thought that the introduction of HAT into a tris-NHC core should provide interesting properties, given the electron-deficient character of this molecule compared to the electron-rich triphenylene (Chart 1).
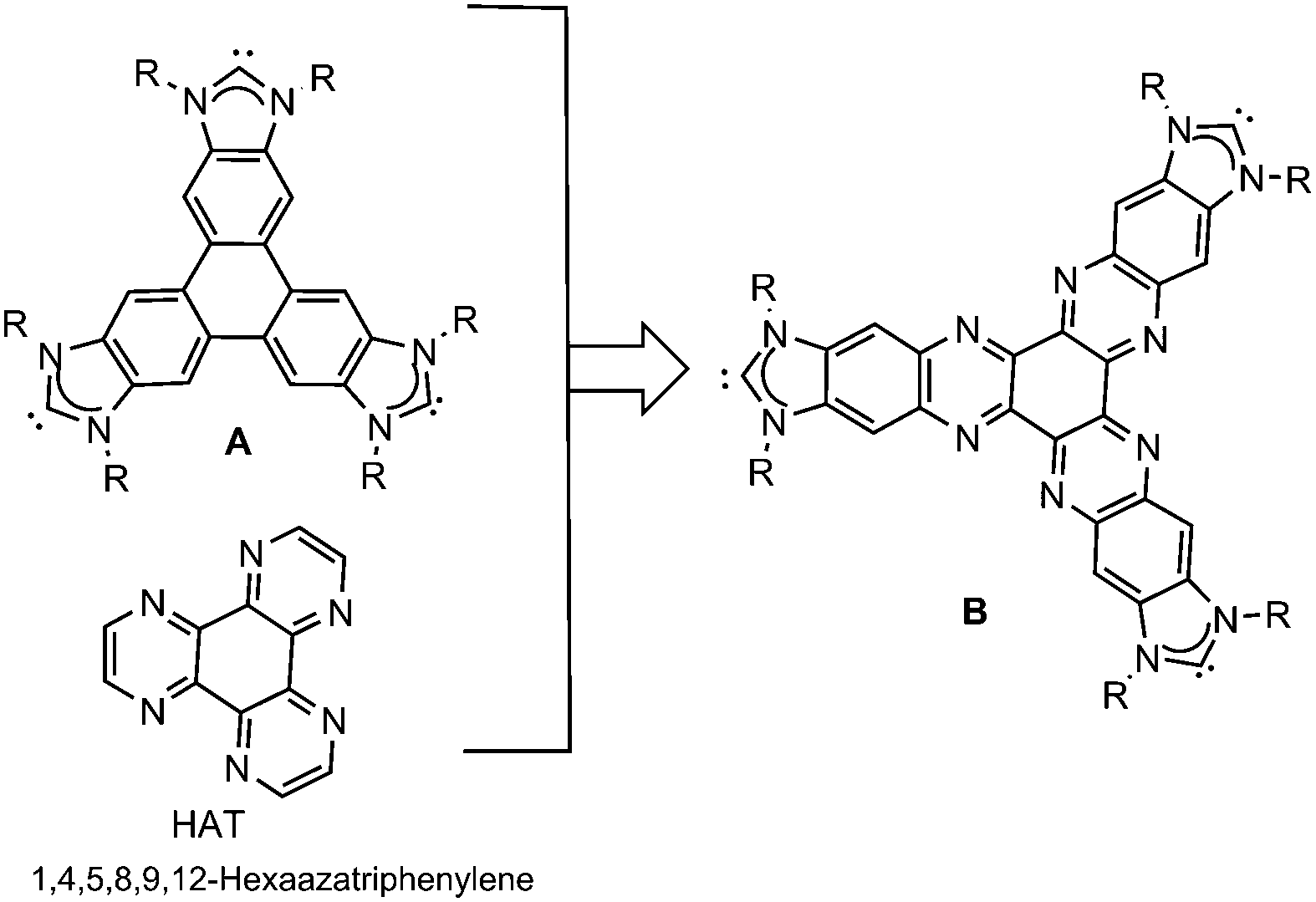 | ||
| Chart 1 | ||
The preparation of the hexaazatriphenylene-tris-benzimidazolium iodide was performed according to the method depicted in Scheme 1. The condensation of 1,3-di-nbutyl-5,6-diaminobenzimidazolium iodide8 with hexaketocyclohexane octahydrate in refluxing glacial acetic acid afforded the tris-azolium iodide [1](I)3 in 81% yield. Anion metathesis of [1](I)3 with [Et3O](BF4) in CH2Cl2 quantitatively afforded the corresponding tetrafluoroborate salt [1](BF4)3. These two salts were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. The elemental analysis of salts [1](I)3 and [1](BF4)3 revealed the presence of three and two water molecules of crystallization, respectively.
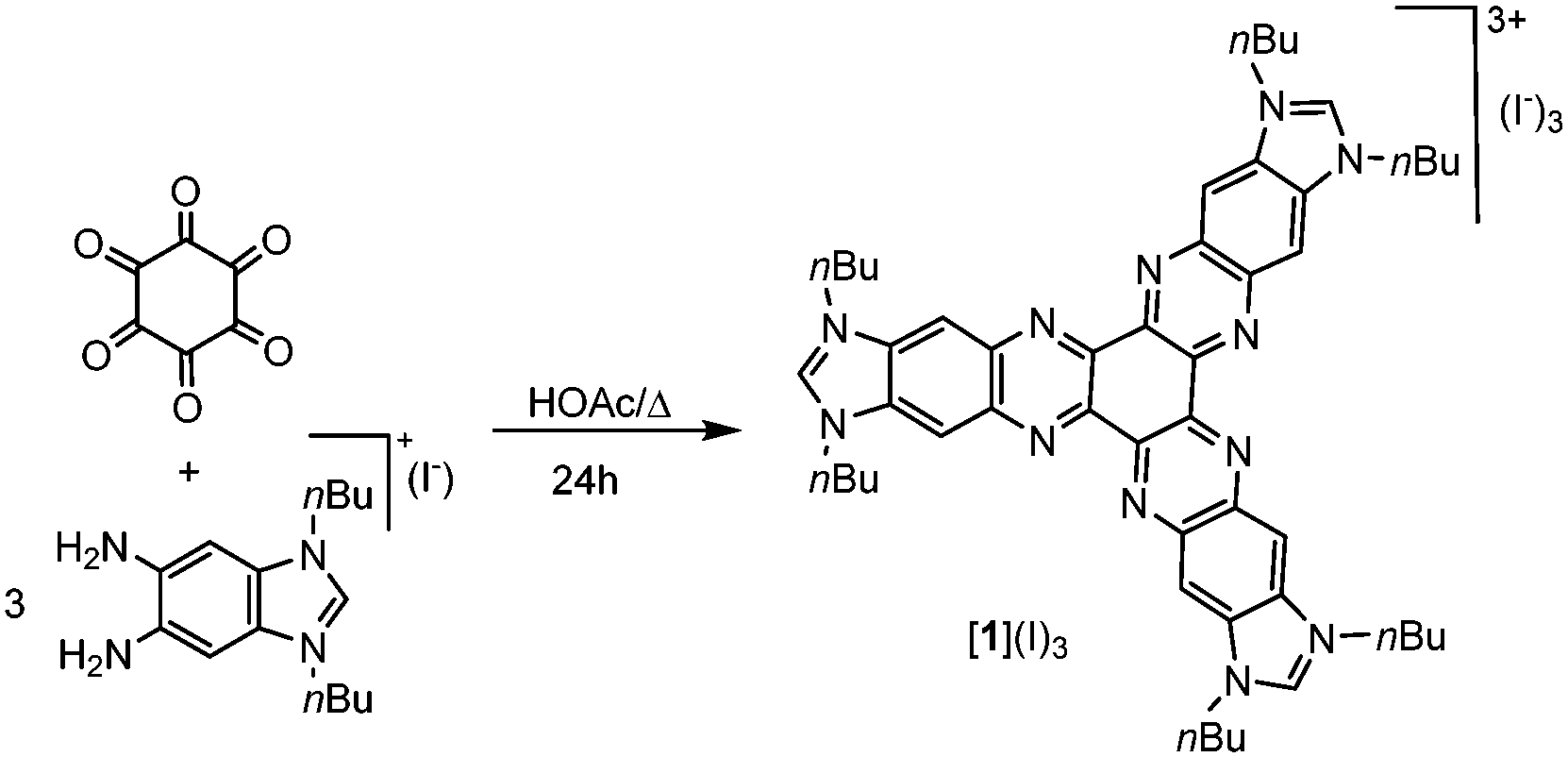 | ||
| Scheme 1 Preparation of [1](I)3. | ||
We sought that [1]3+ should be able to bind to metals through the nitrogen atoms of the hexaazatriphenylene core, and also through the carbon edges of the molecule, if a tris-N-heterocyclic carbene is generated. We first reacted [1](I)3 with [RuCl2(p-cymene)]2, aiming to obtain a compound in which the Ru(p-cymene) fragment could be bound to the ligand through the chelating nitrogen donors. However, instead of obtaining our desired compound, we observed that the reaction afforded [1][RuCl2I(p-cymene)]3, a species in which the iodide from the starting triazolium salt is coordinated to the metal fragment, generating the anionic complex [RuCl2I(p-cymene)]−. All other attempts to achieve the coordination of other metal fragments to the nitrogen atoms of [1]3+ were also unsuccessful. We attributed this behavior to the deactivation of the donor electron pairs at the nitrogen atoms due to the three positive charges of the molecule, and to the high steric strain produced by the presence of the N-butyl groups at the adjacent imidazolium moieties.
The molecular structure of [1][RuCl2I(p-cymene)]3 was further confirmed by single-crystal X-ray diffraction studies (Fig. 1). The determination of the structure of this molecule allowed us to determine the structural parameters of the hexaazatriphenylene-tris-benzimidazolium salt, which may be related to the structure of a potential tris-NHC ligand that could be derived from its deprotonation. The structure consists of three N,N′-dibutylimidazolium fragments linked by a hexaazatriphenylene core. The disposition of the ten fused cycles is essentially coplanar, as can be observed from the lower image of the structure shown in Fig. 1. An interesting feature of the structure is that the distance between the hydrogen atoms of the NCHN fragments of the imidazolium is 15.4 Å. This distance is assumed to be similar to the distance that should be established between the metals of a potential trimetallic complex with a tris-NHC ligand derived from this trisazolium salt.
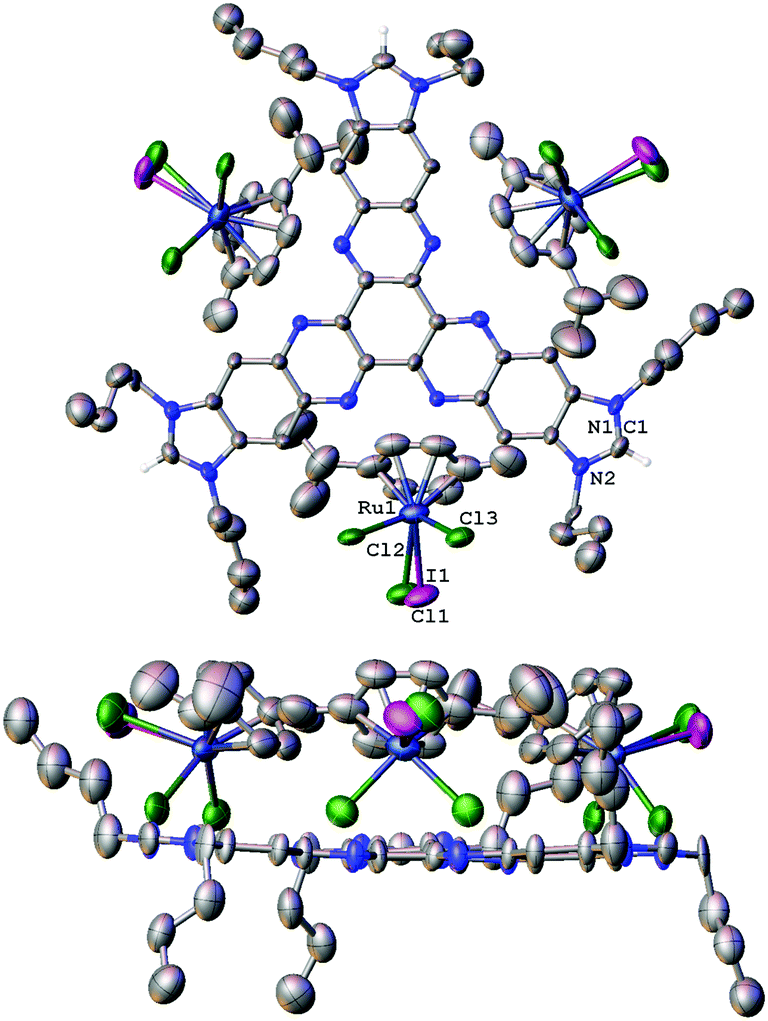 | ||
| Fig. 1 Two perspectives of the molecular structure of [1][RuCl2I(p-cymene)]3. Ellipsoids at 50% probability. Hydrogen atoms and solvents (CH3OH and CH2Cl2) are omitted for clarity of the representation. The crystal contains a mixture of [RuCl3(p-cymene)] and [RuCl2I(p-cymene)] counterions, which were refined with a 40% and 60% occupancy, respectively. Nitrogen atoms represented in blue. | ||
Although all our attempts to coordinate [1]3+ through the nitrogen atoms were unsuccessful, we were able to find a way to use this tricationic compound as a scaffold for the preparation of several planar tris-NHC metal complexes. In all the reactions, the trisazolium salt [1](I)3 was deprotonated by NaOtBu in THF, in the presence of catalytic substoichiometric amounts of NaH at −78 °C (Scheme 2). Then, the metal source was added, and the reaction mixture was allowed to reach room temperature. This method allowed us to obtain complexes 2, 4 and 5 by using [IrCl(COD)]2, [AuCl(SMe)2] and [AuCl(CNC)] (CNC = 2,6-diphenylpyridine), respectively, in low yields ranging from 23–30%. We ascribed these low yields to the formation of the hexaazotriphenylene-tris-benzimidazolidone 6 as a consequence of the deprotonation of the trisazolium [1]3+ and subsequent oxidation of the resulting N-heterocyclic carbene. It is important to mention that the procedures used for the preparation of metal complexes using other known tris-NHC ligands, such as A (Scheme 1), are also very low yielding (yields < 50%), so we did not find the low yields for the coordination using [1](I)3 surprising. The oxidation of NHCs to yield a cyclic urea derivative has been observed by other authors.9 This oxidation process is difficult to prevent because the tricationic salt [1](I)3 is accompanied by a number of water molecules, which come from the hexaketocyclohexane octahydrate used in the preparation of the salt. In fact, the coordination complexes 2, 4 and 5 could be obtained only when the reactions were carried out in the presence of molecular sieves; otherwise, the hexaazatriphenylene-tris-benzimidazolidone 6 was the only product formed. Compound 6 can be isolated from the reaction of [1](I)3 with Ag2O (Scheme 3). The identity of 6 was unequivocally established by single-crystal X-ray diffraction, although the quality of the crystal did not allow us to obtain a molecular structure with the accuracy needed for its publication (see the ESI† for preliminary data and an ORTEP diagram).
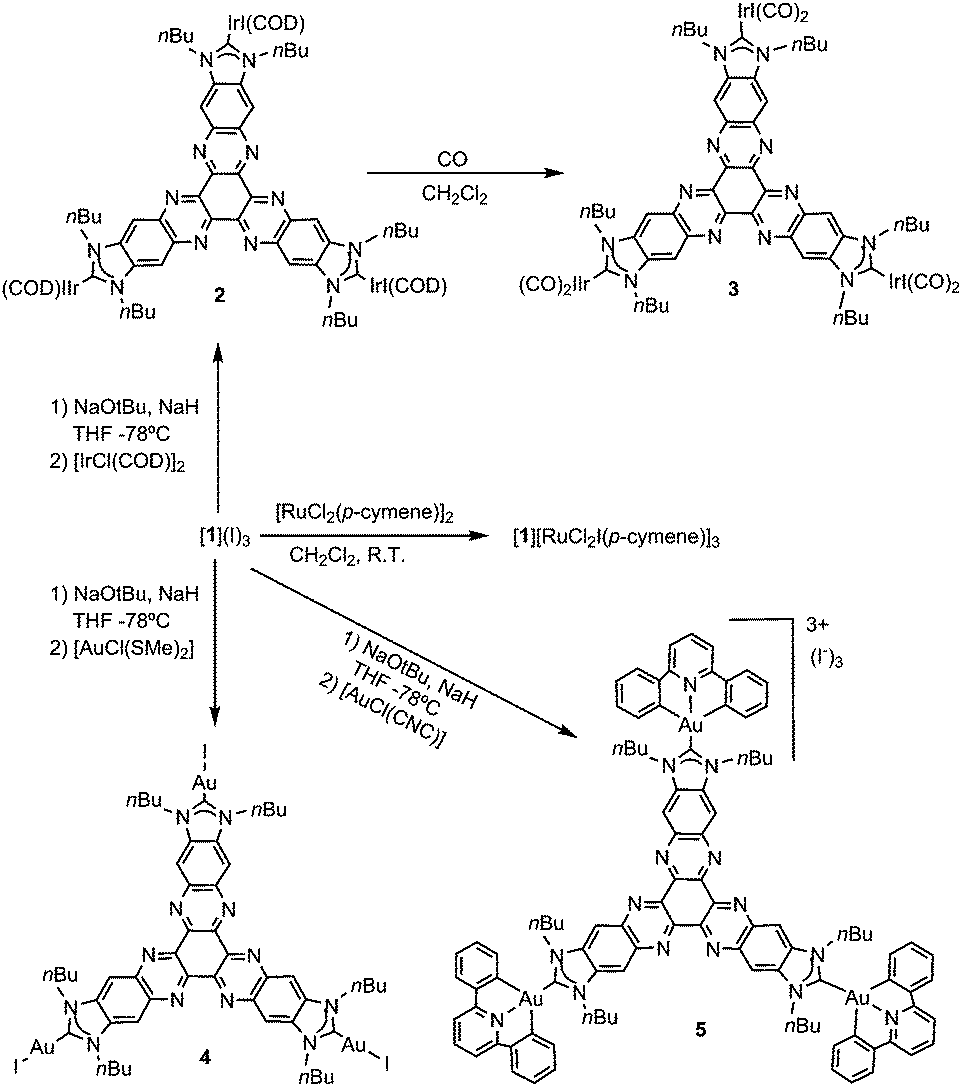 | ||
| Scheme 2 Metal complexes obtained in this work. | ||
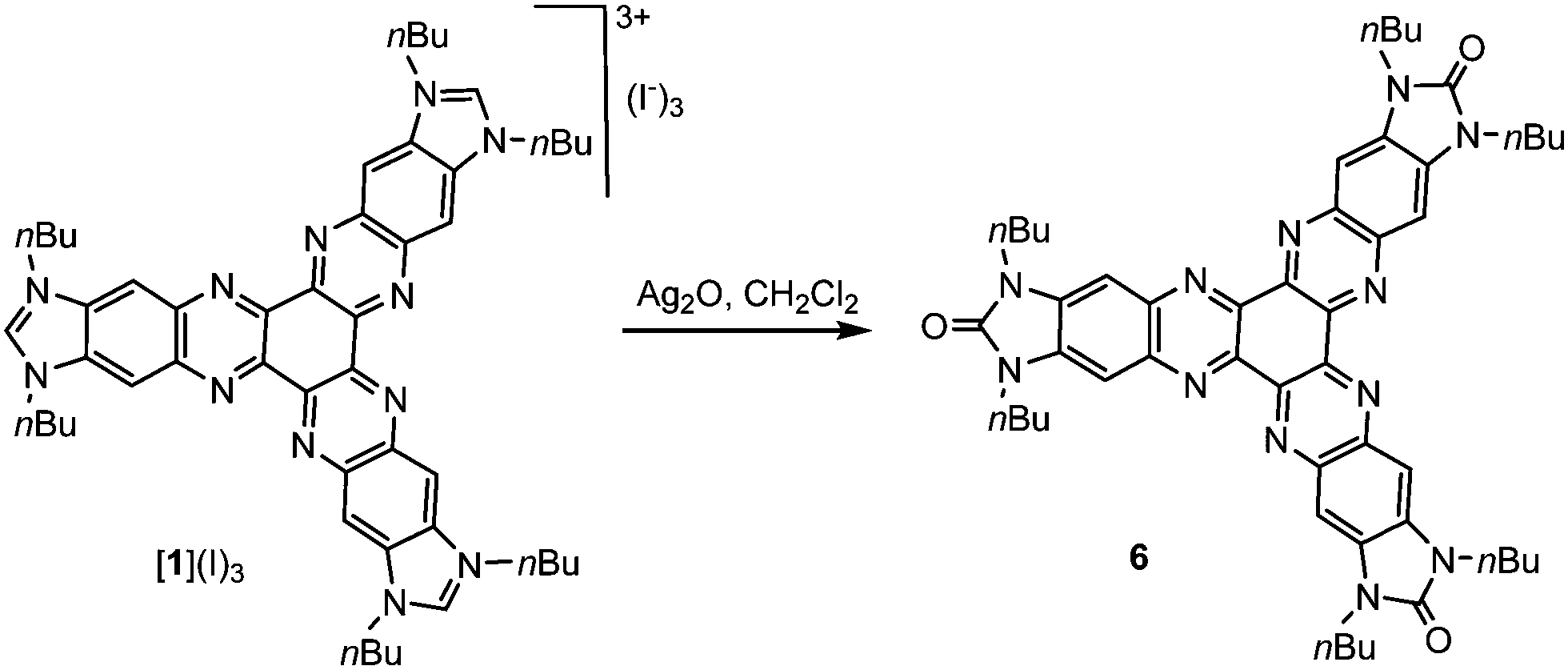 | ||
| Scheme 3 Preparation of compound 6. | ||
All new complexes were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. Unfortunately, we were unable to obtain single crystals of the quality required to allow the determination of any of their molecular structures by X-ray diffraction. As a common feature, the 1H NMR spectra of 2, 4 and 5 (and also 6) were consistent with the threefold symmetry of their predicted geometries. Their 13C NMR spectra revealed the signals due to the metallated carbene carbons at 205.6, 197.2 and 172.3 ppm, for 2, 4 and 5, respectively. These signals are significantly downfield shifted compared to those of other related NHC-based complexes of iridium(I), gold(I) and gold(III), an observation that may be related to the low electron-donating character of the tris-NHC ligand.10
The reaction of 2 with carbon monoxide in CH2Cl2 afforded the hexacarbonylated complex 3. This compound displayed two IR bands at 2069 and 1992 cm−1, assigned to the CO stretching vibrations. The comparison of these values with those given by related NHC–IrI(CO)211 and NHC–IrCl(CO)212 complexes indicates that the electron-donating character of the tris-NHC ligand in 3 is significantly reduced, as expected by the presence of the electron-poor hexaazatriphenylene core.
In order to determine if the new tris-NHC ligand may have applications in homogeneous catalysis, we decided to test the tris–Au(I) complex 4 in two benchmark reactions. We chose this compound for our preliminary studies because the activity of gold-based catalysts has often been related to the ability of the Au center to behave as an electrophile. Given the low electron-donating character of the tris-NHC ligand in 4, we thought that this complex should yield good catalytic efficiencies. We first tested complex 4 in the hydroamination of terminal alkynes,13 a reaction for which several Au(I) complexes have shown good activity.6a,14 In fact, this reaction has been used as a model for testing the effects of modifying the Lewis acidity of the gold centre on its catalytic activity, and the studies demonstrated that increasing the Lewis acid character of the metal results in enhanced activity.11,14h,fTable 1 summarizes the results that we obtained for the hydroamination of phenylacetylene with five different primary amines. The reactions were carried out in acetonitrile at 90 °C, using a catalyst loading of 0.33 mol% (1 mol% based on the amount of active gold centres). AgBF4 was added as a chloride scavenger in order to activate the catalyst. We also tested the catalytic activity of the complex [AuCl(1,3-di-nbutyl-benzimidazolylidene)], 7.15 Complex 7 can be considered as one of the gold-containing branches of 4, but lacking the electron-poor pyrazine ring. This makes 7 a perfect pattern for comparison because it bears a ligand with the same steric properties as the triscarbene ligand in 4, but with a stronger electron-donating character.12b As seen from the results shown in Table 1, the activity of 4 was excellent for all the combinations of substrates used, with product yields ranging from 81–98%. On the contrary, the activity of the monometallic complex 7 was only moderate, affording product yields 14–20% points below those given by 4. This result illustrates how the reduction of the electron-donating character of the NHC ligand, produced by the presence of the HAT core, has a positive impact on the catalytic activity of 4. All in all, compound 4 is less active than the best Au(I) catalysts reported to date for this reaction.14j
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Ar | Yieldb |
| a Reaction conditions: 0.5 mmol phenylacetylene, 0.55 mmol amine, 1% mol catalyst (based on concentration of Au active sites), 2% mol AgBF4, 1 mL of MeCN at 90 °C, 6 h. b Yields determined by GC using anisole as internal standard. | |||
| 1 | 4 | Ph | 92 |
| 2 | 7 | Ph | 72 |
| 3 | 4 | 2-MeC6H4 | 98 |
| 4 | 7 | 2-MeC6H4 | 80 |
| 5 | 4 | 4-MeC6H4 | 91 |
| 6 | 7 | 4-MeC6H4 | 72 |
| 7 | 4 | 2,4,6-Me3C6H2 | 82 |
| 8 | 7 | 2,4,6-Me3C6H2 | 68 |
| 9 | 4 | 2,6-iPr2C6H3 | 81 |
| 10 | 7 | 2,6-iPr2C6H3 | 64 |
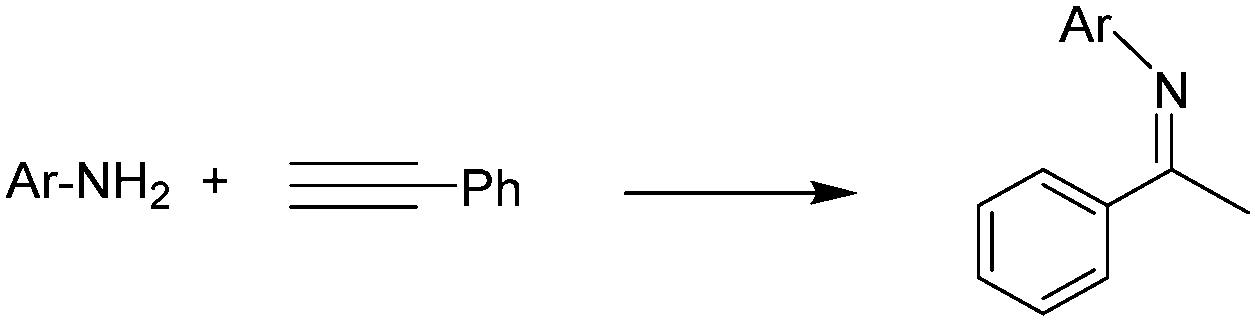
We also tested 4 in the three-component Strecker reaction for the synthesis of α-aminonitriles by reaction of an amine, an aldehyde (or ketone) and trimethylsilyl cyanide.16 For comparative purposes, we also tested the activity of the benzimidazolylidene–Au(I) complex 7. To our knowledge, there is only one example of a Au(I) complex used to promote this process.7 This reaction is normally catalyzed by complexes with Lewis acid functions, so we thought that 4 may be able to promote this process. The reactions were carried out at room temperature in CH2Cl2 using a catalyst loading of 4 mol% (based on the amount of Au(I) active centers). As can be seen in Table 2, the results are highly dependent on the combination of the substrates used. For the reactions carried out with acetophenone, 4-bromoacetophenone and 4-methoxyacetophenone, 4 provided excellent yields, ranging from 78–99%, with the highest activity shown for the reaction of 4-bromoacetophenone. This observation indicates that the catalyst tolerates the presence of halides in the substrate, thus affording a clear advantage over palladium-based catalysts,17 for which the dehalogenation of the halide-substituted ketone may constitute an important side reaction. The product yield was very low for the reaction carried out using p-nitroacetophenone (22%), but it has to be taken into account that this substrate is highly deactivated for the coupling with aniline, thus rarely providing yields over 20%. As seen from the data shown in Table 2, the activity of the monometallic complex 7 is significantly lower, thus indicating that the presence of the HAT core in the trimetallic complex 4 improves the activity of the complex.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | R | Yieldb |
| a Reaction conditions: 0.5 mmol ketone, 0.55 mmol aniline, 1 mmol TMSCN, 4% mol catalyst loading (based on the concentration of Au active sites), 2 mL of CH2Cl2, 24 h at room temperature. b GC yields using anisole as internal standard. | |||
| 1 | 4 | H | 77 |
| 2 | 7 | H | 39 |
| 3 | 4 | Br | 99 |
| 4 | 7 | Br | 59 |
| 5 | 4 | NO2 | 22 |
| 6 | 7 | NO2 | 5 |
| 7 | 4 | MeO | 79 |
| 8 | 7 | MeO | 52 |
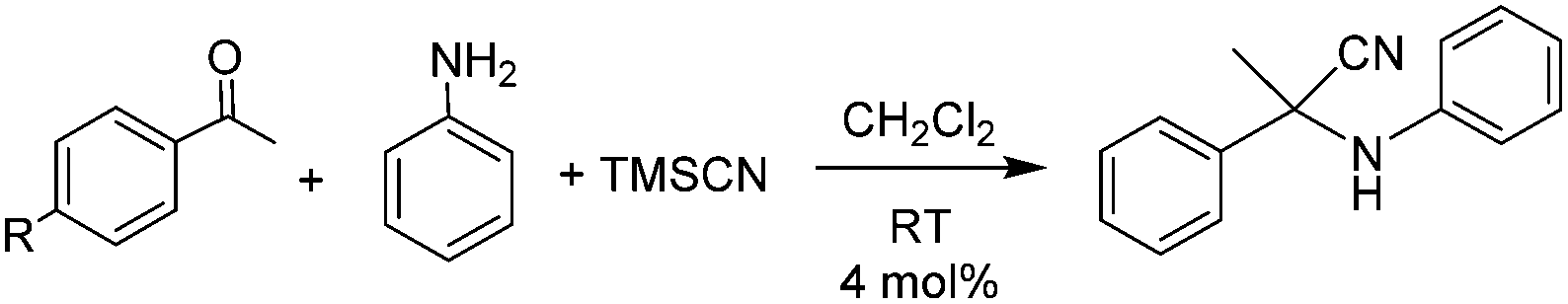
In summary, we prepared a new hexaazatriphenylene-tris-benzimidazolium salt, a precursor of a planar hexaazatriphenylene-tris-benzimidazolylidene ligand. The ligand was coordinated to several metal fragments, allowing the preparation of two iridium and two gold star-shaped tris-NHC complexes. The catalytic activity of the tris-NHC–Au(I) complex was tested in the hydroamination of phenylacetylene and in the three-component Strecker reaction, where it displayed excellent activities due to the presence of the hexaazatriphenylene linker in the ligand, which enhances the electrophilic character of the gold centers in the catalyst.
We gratefully acknowledge financial support from MINECO of Spain (CTQ2014-51999-P) and the Universitat Jaume I (P11B2014-02 and P11B2015-24). We are grateful to the Serveis Centrals d'Instrumentació Científica (SCIC-UJI) for providing the spectroscopic facilities.
Notes and references
- J. L. Segura, R. Juarez, M. Ramos and C. Seoane, Chem. Soc. Rev., 2015, 44, 6850–6885 RSC.
- S. Kitagawa and S. Masaoka, Coord. Chem. Rev., 2003, 246, 73–88 CrossRef CAS.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed.
- E. Peris, Chem. Commun., 2016, 52, 5777–5787 RSC.
- E. Peris, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00695.
- (a) S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2013, 52, 7009–7013 CrossRef CAS PubMed; (b) S. Gonell, R. G. Alabau, M. Poyatos and E. Peris, Chem. Commun., 2013, 49, 7126–7128 RSC.
- S. Gonell, M. Poyatos and E. Peris, Chem. – Eur. J., 2014, 20, 5746–5751 CrossRef CAS PubMed.
- D. Tapu, Z. McCarty, L. Hutchinson, C. Ghattas, M. Chowdhury, J. Salerno and D. VanDerveer, J. Organomet. Chem., 2014, 749, 134–141 CrossRef CAS.
- (a) M. Schmidtendorf, J. Organomet. Chem., 2014, 751, 620–627 CrossRef CAS , C. S. to Brinke and F. E. Hahn; (b) M. Slivarichova, R. Ahmad, Y. Y. Kuo, J. Nunn, M. F. Haddow, H. Othman and G. R. Owen, Organometallics, 2011, 30, 4779–4787 CrossRef CAS; (c) A. Petronilho, H. Muller-Bunz and M. Albrecht, Chem. Commun., 2012, 48, 6499–6501 RSC.
- (a) Q. Teng and H. Han Vinh, Inorg. Chem., 2014, 53, 10964–10973 CrossRef CAS PubMed; (b) J. C. Bernhammer and H. V. Huynh, Dalton Trans., 2012, 41, 8600–8608 RSC.
- S. Ibañez, M. Poyatos, L. N. Dawe, D. Gusev and E. Peris, Organometallics, 2016, 35, 2747–2758 CrossRef.
- (a) H. Valdes, M. Poyatos and E. Peris, Organometallics, 2014, 33, 394–401 CrossRef CAS; (b) H. Valdes, M. Poyatos and E. Peris, Inorg. Chem., 2015, 54, 3654–3659 CrossRef CAS PubMed; (c) A. R. Chianese, X. W. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 CrossRef CAS; (d) R. A. Kelly, III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef; (e) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- L. Huang, M. Arndt, K. Goossen, H. Heydt and L. J. Goossen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- (a) M. J. Pouy, S. A. Delp, J. Uddin, V. M. Ramdeen, N. A. Cochrane, G. C. Fortman, T. B. Gunnoe, T. R. Cundari, M. Sabat and W. H. Myers, ACS Catal., 2012, 2, 2182–2193 CrossRef CAS; (b) M. Katari, M. N. Rao, G. Rajaraman and P. Ghosh, Inorg. Chem., 2012, 51, 5593–5604 CrossRef CAS PubMed; (c) L. Canovese, F. Visentin, C. Levi and C. Santo, Inorg. Chim. Acta, 2012, 391, 141–149 CrossRef CAS; (d) E. Alvarado, A. C. Badaj, T. G. Larocque and G. G. Lavoie, Chem. – Eur. J., 2012, 18, 12112–12121 CrossRef CAS PubMed; (e) S. Gaillard, J. Bosson, R. S. Ramon, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2010, 16, 13729–13740 CrossRef CAS PubMed; (f) H. Yang and F. P. Gabbai, J. Am. Chem. Soc., 2015, 137, 13425–13432 CrossRef CAS PubMed; (g) X. Hu, D. Martin and G. Bertrand, New J. Chem., 2016, 40, 5993–5996 RSC; (h) P. Barrio, M. Kumar, Z. Lu, J. Han, B. Xu and G. B. Hammond, Chem. – Eur. J., 2016, 22, 16410–16414 CrossRef CAS PubMed; (i) Y. Wang, Z. Wang, Y. Li, G. Wu, Z. Cao and L. Zhang, Nat. Commun., 2014, 5, 3470 Search PubMed; (j) D. Malhotra, M. S. Mashuta, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 4456–4459 CrossRef CAS PubMed.
- M. C. Jahnke, J. Paley, F. Hupka, J. J. Weigand and F. E. Hahn, Z. Naturforsch., 2009, 64, 1458–1462 CAS.
- (a) J. Jarusiewicz, Y. Choe, K. S. Yoo, C. P. Park and K. W. Jung, J. Org. Chem., 2009, 74, 2873–2876 CrossRef CAS PubMed; (b) S. J. Connon, Angew. Chem., Int. Ed., 2008, 47, 1176–1178 CrossRef CAS PubMed; (c) C. Spino, Angew. Chem., Int. Ed., 2004, 43, 1764–1766 CrossRef CAS PubMed; (d) H. Groger, Chem. Rev., 2003, 103, 2795–2827 CrossRef PubMed; (e) L. Yet, Angew. Chem., Int. Ed., 2001, 40, 875–877 CrossRef CAS; (f) A. Strecker, Ann. Chem. Pharm., 1850, 75, 27–45 CrossRef.
- (a) Q. Q. Teng and H. V. Huynh, Inorg. Chem., 2014, 53, 10964–10973 CrossRef CAS PubMed; (b) J. Choi, H. Y. Yang, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2010, 49, 7718–7722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of new compounds, CIF file of the molecular structure of [1][RuCl2I(p-cymene)]3 and a summary of the crystal data, data collection and refinement details. CCDC 1528547. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc00525c |
| This journal is © The Royal Society of Chemistry 2017 |