
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular anharmonicity in molecular crystals: interplay between experimental low-frequency dynamics and quantum quasi-harmonic simulations of solid purine†
Michael T.
Ruggiero
 *a,
J. Axel
Zeitler
a and
Alessandro
Erba
b
*a,
J. Axel
Zeitler
a and
Alessandro
Erba
b
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK. E-mail: mtr34@cam.ac.uk
bDipartimento di Chimica, Universitá di Torino, via Giuria 5, Torino, Italy
First published on 9th March 2017
Abstract
The intermolecular anharmonic potential of crystalline purine is probed by means of temperature-dependent terahertz time-domain spectroscopy, low-frequency Raman scattering, X-ray diffraction, and ab initio quasi-harmonic quantum-chemical simulations. As temperature increases, anharmonicity in the intermolecular interactions results in strongly anisotropic thermal expansion – with a negative thermal expansion along the b crystallographic axis – yielding corresponding bulk structural modifications. The observed thermally-induced shifts of most vibrational bands in the terahertz region of the spectra are shown to arise from volume-dependent thermal changes of the hydrogen-bond pattern along the a and b crystallographic axes.
Molecular crystals are increasingly being used for a variety of applications, such as high energy-density media, active pharmaceutical ingredients (APIs), and components of linear and non-linear optical devices.1–3 Crystalline materials are often described from two distinct perspectives, either in their static structural or in their dynamic vibrational aspects.4,5 While these generalised pictures have both proven to be valid and accurate for rationalising and predicting some properties of solids independently from one another, they should obviously be considered as complementary in the overall understanding of many of their features.6–8 From a fundamental standpoint both phenomena are a result of the shape of the potential energy surface (PES), with the PES minima and curvature governing equilibrium structures and dynamics, respectively. Experimentally, it is possible to characterise the PES-dependent effects using a variety of techniques, for example X-ray diffraction for structures and vibrational spectroscopy for dynamics.9
In particular, when molecular crystals are considered, the characterisation of their relatively flat PES proves particularly delicate and yet is key to the understanding of their structural, mechanical, and thermodynamic properties, which govern their rich polymorphism, for example.10 In this respect, low-frequency vibrational methods such as terahertz time-domain spectroscopy (THz-TDS) and Raman scattering would constitute powerful tools for probing both the intra- and inter-molecular PES of such systems, since the relevant vibrations mostly involve motions of entire molecules, rather than only individual bonds.11,12 However, the interpretation of the low-frequency spectra is far more complicated than it is for their mid-infrared counterparts, since the corresponding vibrations strongly depend on the intermolecular interactions, thus implying that each material has a unique spectral fingerprint at THz frequencies.13,14 In this regard, the complementary use of ab initio simulations is nowadays commonly acknowledged as necessary in order to fully understand the experimental results,10,15–17 provided that they are highly-accurate in their description of weak intermolecular forces.18–20
Further complicating the analysis of molecular crystals is the presence of anharmonicity in the PES, a common phenomenon that strongly influences the bulk properties of materials, especially those that depend on temperature.21,22 While this effect is almost always present in solids it becomes especially pronounced in the case of weakly-bonded molecular crystals.23 This leads to the properties of molecular crystals being significantly influenced even by modest temperature changes close to ambient conditions; understanding their thermal properties is thus of critical importance for most potential applications.24 But while these effects can be observed experimentally, the theoretical interpretation is made much more difficult since density functional theory (DFT) calculations are typically performed at absolute zero and vibrational dynamics are often calculated within the harmonic approximation, which, by definition, neglects anharmonic terms.25–27 An effective way of accounting for some anharmonic effects of solids (such as thermal expansion, temperature-dependent elastic response, etc.) within standard DFT is represented by the quasi-harmonic approximation (QHA), which requires the evaluation of lattice dynamics at different volumes and thus enables the description of volume-dependent thermal features.28,29 Only very recently has this methodology been used to investigate the thermal properties of molecular crystals,30–33 and is here applied for the first time to interpret the temperature-dependent low-frequency spectral properties of a molecular solid.
Recently, it was shown that crystalline purine, an important component of DNA bases, exhibits significant changes in its THz-TDS spectrum as a function of temperature, strongly indicating that there exists significant intermolecular anharmonicity.34,35 The origin of such strong anharmonic features is explored here, by performing variable temperature THz-TDS measurements, low-frequency Raman scattering experiments, and quantum-mechanical quasi-harmonic simulations. Samples of crystalline purine were used as received (Sigma-Aldrich, >98%) and were either mixed with high-density polyethylene (10% w/w) for THz-TDS or simply ground for the Raman measurements. The THz-TDS data were acquired on a commercial TeraView Terapulse 4000 spectrometer from 20–90 cm−1, and the low-frequency Raman data utilized a Horiba LabRAM HR Evolution spectrometer operating at 532 nm coupled with an ultra-low frequency accessory from 25–150 cm−1, each having a spectral resolution of 1 cm−1. The results of these experiments, shown in Fig. 1, clearly demonstrate that, with the exception of the feature occurring at 46.5 cm−1 in both spectra, all of the peaks sharpen and shift to higher frequencies upon cooling to great effect.
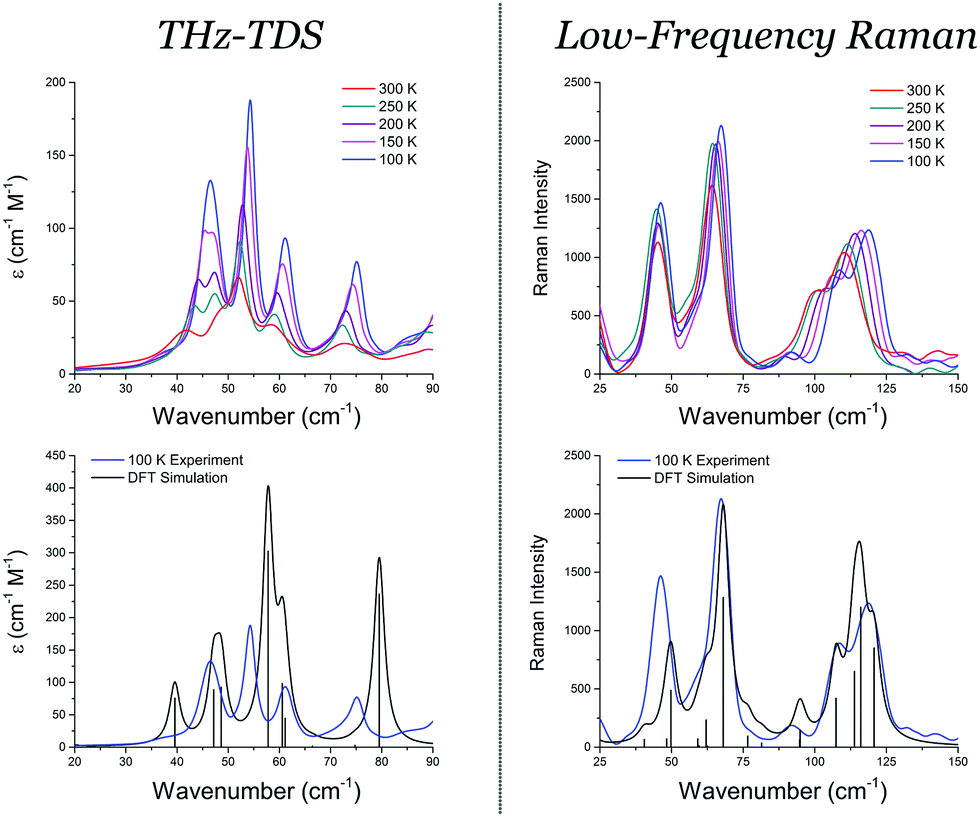 | ||
| Fig. 1 Variable temperature THz-TDS and low-frequency Raman of crystalline purine from 100 K to 300 K in 50 K intervals. The bottom panels show the simulated vibrational spectra (scaled by 0.85) as determined by equilibrium solid-state DFT calculations compared to the 100 K experimental spectra. | ||
Solid-state DFT simulations were performed in order to assign the spectra and determine the nature of the vibrational features. A developmental version of the Crystal14 software package was used for all simulations.36 The atoms were modeled using the all-electron def2-SVP basis set37 and the Perdew–Burke–Ernzerhof (PBE) density functional,38 and corrected for dispersion forces by means of the D3 correction by Grimme.39 The crystal structure of purine was fully-optimised with no constraints other than those given by the orthorhombic space group it belongs to (Pna21) prior to the simulation of vibrational properties, and the IR and Raman intensities were calculated analytically via a Coupled-Perturbed Kohn–Sham (CPKS) method,41–43 both to a convergence of ΔE < 10−10 Hartree (additional details available in the ESI†). The results of the vibrational simulation are shown in Fig. 1. As previously reported, the simulation slightly overestimates the low-frequency vibrational features, requiring a scaling of 0.85 in order to come into agreement with the experiment, and this factor was used for all predicted vibrational frequencies discussed here.35
In order to shed some light on the origin of the thermally-induced shifts of the low-frequency peaks, advantage has been taken of a recently developed quasi-harmonic algorithm for lattice dynamical calculations, as implemented in the Crystal software.44–48 Vibrational frequencies are computed at four volumes (from a −2% compression to a +4% expansion with respect to the static equilibrium volume) after full volume-constrained structural relaxations, which provide an accurate description of anisotropic thermally-induced structural changes. The results of the QHA simulations constitute a means of correlating structural modifications with temperature and thus of predicting thermally-induced shifts of spectral peaks as well. The computed frequencies and shifts (consistently scaled by 0.85) are reported in Table 1, along with the corresponding experimental IR and Raman ones. The absolute magnitude of the measured shifts between 300 K and 100 K (0.6, 1.6, 1.7, 2.2, 2.5, 5.1, 9.0 and 9.1 cm−1) is such to represent a formidable challenge to the DFT simulations in order to be reliably reproduced. Indeed, let us stress that a shift by 1 cm−1 corresponds to an energy difference of 0.003 kcal mol−1 (more than 300 times smaller than the very much sought for “chemical accuracy” in the theoretical chemistry field).39,40 Very tight numerical parameters, combined with the robustness of the adopted algorithms, have been used (energy convergence threshold for each atomic displacement set to 10−10 Hartree and a reciprocal space sampling over 216 k points in the irreducible Brillouin zone) to attempt the first such characterisation. All of the vibrational frequencies are blue-shifted upon cooling, which is reproduced by the QHA simulations. The predicted shifts match the experimentally-measured ones to a large extent for most peaks, particularly so in terms of relative amplitudes, which is a remarkable achievement if the required numerical accuracy is considered, as discussed above. A partial exception to this agreement are the IR modes occurring at 46.5 and 75.9 cm−1 in the 100 K spectrum, which have their shifts underestimated and overestimated (as well as their predicted intensities), respectively. Both modes are indeed believed to exhibit some intrinsic vibrational anharmonicity (not accounted for at the QHA level of theory); previous studies have reported that both features continue to sharpen and shift significantly upon cooling to 4 K, unlike the other absorptions that show little change below 100 K.34,35 Further accentuating the intrinsic vibrational anharmonicity of the 75.9 cm−1 feature is the asymmetry of the vibrational band compared to the others, which is a strong indicator of this particular phenomenon.49
| Peak position (cm−1) | Shifts (cm−1) | |||||
|---|---|---|---|---|---|---|
| Exp. | QHA | Exp. | QHA | Exp. | QHA | |
| 300 K | 300 K | 100 K | 100 K | |||
| IR | 41.4 | 46.3 | 46.5 | 48.0 | 5.1 | 1.7 |
| IR | 47.1 | 49.1 | 46.5 | 50.3 | 0.6 | 1.2 |
| Raman | 46.4 | 46.4 | 0.0 | |||
| IR | 52.0 | 56.9 | 54.2 | 59.5 | 2.2 | 2.6 |
| IR | 59.3 | 61.7 | 61.0 | 62.2 | 1.7 | 0.5 |
| Raman | 66.2 | 67.6 | 67.8 | 68.2 | 1.6 | 0.6 |
| IR | 73.4 | 77.2 | 75.9 | 81.4 | 2.5 | 4.2 |
| Raman | — | 89.3 | 92.1 | 94.6 | — | 5.3 |
| Raman | 99.5 | 101.1 | 108.6 | 107.2 | 9.1 | 6.1 |
| Raman | 110.5 | 109.7 | 119.5 | 116.0 | 9.0 | 6.3 |
The QHA frequency shifts can be correlated with modifications of particular structural groups, by examination of the corresponding normal mode displacement vectors. Low-frequency vibrational features of solid purine are dominated by motion of the intermolecular hydrogen bonds, lying in the (ab) plane. Indeed, there is a clear correlation between the relative amount of frequency shifting and hydrogen bond perturbation, particularly so in the N–H⋯H bond angle rather than bond length (Fig. 2). Both Table 1 and Fig. 2 confirm that a very large fraction of the observed temperature-dependent shifts of the low-frequency intermolecular spectral features of solid purine are due to thermally-induced structural changes rather than to intrinsic vibrational anharmonicity. A deeper investigation of the crystal structure of purine as a function of temperature is then needed, both experimentally and theoretically.
 | ||
| Fig. 2 The change in amine hydrogen bond angle (top) and bond distance (bottom) for each low-frequency normal mode determined from the equilibrium DFT simulation (solid line). The amount of QHA thermally-induced shift is also shown (dashed line). Experimentally active vibrational bands are marked with blue stars and red diamonds for IR and Raman, respectively. | ||
Quasi-harmonic lattice dynamics represents an effective way to determine the temperature dependence of structural and elastic properties of solids. Volumetric thermal expansion and temperature-dependent bulk modulus of solid purine are reported in the two bottom-right panels of Fig. 3. The sole inclusion of the zero-point vibrational effect results in a volume increase of 0.8% and a bulk modulus decrease of 10%, with respect to the static 0 K equilibrium DFT values. Thermal effects at 300 K further increase the volume by 1.2% and decrease the bulk modulus by 12% (2% and 20% with respect to the static 0 K DFT values, respectively). From these figures, it clearly emerges that even slight structural changes are dramatically affecting the mechanical properties of solid purine. In order to better highlight how the structure is affected by temperature, the thermal expansion of purine is reported in panel A of Fig. 3, as determined from the QHA simulation, which is found to be rather anisotropic with a large expansion along the c axis (where molecules are mainly held together by weak dispersive interactions), a smaller expansion along the a axis, and a small negative thermal expansion along the b axis (where stronger hydrogen-bond interactions are taking place).
 | ||
| Fig. 3 Results of the QHA simulations, showing (A) the thermal expansion coefficient of each crystal axis, (B, top) the predicted lattice constants as a function of temperature (with experimental values presented as points), (B, middle) the predicted volume as a function of temperature, (B, bottom) the bulk modulus as a function of temperature, and (C) the optimised 0 K crystal structure. | ||
From an experimental point of view, single crystals of purine were grown via sublimation of the as-received sample, and SC-XRD data obtained at three temperatures (100 K, 200 K, and 300 K)50,51 (additional details provided in the ESI†). The data confirm the overall QHA picture, with small structural changes to a and b (with the b axis showing a slight contraction), and with the c axis exhibiting the greatest amount of expansion. As regards absolute values, theoretical lattice parameters at 100 K turn out to be slightly underestimated by −0.27%, −2.15%, and −0.63% in the a, b, and c axes, respectively. The overall computed contraction of the unit cell may partially explain the need for the applied scaling factor to the vibrational frequencies.
The SC-XRD and QHA data enable a precise atomic-level analysis of the temperature-dependent structure of solid purine. It is clear that small thermally-induced structural changes largely affect the strength of the intermolecular interactions, which is reflected in large mechanical softening and spectral peak shifting. Intermolecular interactions along the a axis produce no relevant structural modifications other than an expansion, which linearly follows the corresponding lattice parameter. The analysis of the relative orientation of the molecules with respect to one another (for example the angle between the hydrogen-bonded molecular sheets), rather than simply of the distances between them, provides insight. The angle formed by the molecules and the (ab) plane, (001) Miller plane, opens by 1.77° upon cooling, while the inter-chain distance simultaneously contracts (Fig. 4). Here, the contraction of the hydrogen bond is negated by the change in the packing angle, leading to the observed negative thermal expansion behaviour.
 | ||
| Fig. 4 View down the a crystallographic axis showing two of the major thermally-induced structural changes in solid purine. | ||
This work shows how some anharmonic thermal features of molecular crystals can effectively be understood at an atomistic level by coupling quasi-harmonic lattice-dynamical simulations with experimental THz-TDS, Raman scattering, and single crystal X-ray diffraction. The success of the present investigation suggests that quasi-harmonic simulations could provide a reliable description of the thermo-mechanical and thermodynamic properties of this class of weakly-bound solids.
M. T. R. and J. A. Z. thank the UK Engineering and Physical Sciences Research Council for funding (EP/N022769/1). M. T. R. also thanks the European Molecular Biology Organization for travel funding. We also thank Sara Dampf and Adam Zaczek for their assistance with crystallisation.
References
- O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- T. Beyer, G. M. Day and S. L. Price, J. Am. Chem. Soc., 2001, 123, 5086–5094 CrossRef CAS PubMed.
- L. E. Fried, M. R. Manaa, P. F. Pagoria and R. L. Simpson, Annu. Rev. Mater. Sci., 2001, 31, 291–321 CrossRef CAS.
- A. Rousse, C. Rischel and J.-C. Gauthier, Rev. Mod. Phys., 2001, 73, 17 CrossRef CAS.
- T.-A. V. Khuong, J. E. Nuñez, C. E. Godinez and M. A. Garcia-Garibay, Acc. Chem. Res., 2006, 39, 413 CrossRef CAS PubMed.
- E. T. J. Nibbering and T. Elsaesser, Chem. Rev., 2004, 104, 1887–1914 CrossRef CAS PubMed.
- W. T. M. Mooij, B. P. van Eijck and J. Kroon, J. Am. Chem. Soc., 2000, 122, 3500–3505 CrossRef CAS.
- B. P. van Eijck, J. Comput. Chem., 2001, 22, 816–826 CrossRef CAS.
- N. Mounet and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 205214 CrossRef.
- G. Beran, Chem. Rev., 2016, 116, 5567 CrossRef CAS PubMed.
- C. A. Schmuttenmaer, Chem. Rev., 2004, 104, 1759–1779 CrossRef CAS PubMed.
- E. P. J. Parrott and J. A. Zeitler, Appl. Spectrosc., 2015, 69, 1–25 CrossRef CAS PubMed.
- S. X. Yin, S. P. Delaney, T. M. Smith, T. M. Korter, D. Pan and T. M. Korter, Cryst. Growth Des., 2013, 13, 2943–2952 Search PubMed.
- M. T. Ruggiero, J. Sibik, J. A. Zeitler and T. M. Korter, J. Phys. Chem. A, 2016, 120, 7490–7495 CrossRef CAS PubMed.
- M. D. King, W. D. Buchanan and T. M. Korter, Phys. Chem. Chem. Phys., 2011, 13, 4250 RSC.
- T. R. Juliano, M. D. King and T. M. Korter, IEEE Trans. Terahertz Sci. Technol., 2013, 3, 281 CrossRef CAS.
- M. R. C. Williams, D. J. Aschaffenburg, B. K. Ofori-Okai and C. A. Schmuttenmaer, J. Phys. Chem. B, 2013, 117, 10444 CrossRef CAS PubMed.
- S. Wen, K. Nanda, Y. Huang and G. J. O. Beran, Phys. Chem. Chem. Phys., 2012, 14, 7578–7590 RSC.
- J. Moellmann and S. Grimme, J. Phys. Chem. C, 2014, 118, 7615–7621 CAS.
- M. A. Neumann and M.-A. Perrin, J. Phys. Chem. B, 2005, 109, 15531–15541 CrossRef CAS PubMed.
- R. G. Della Valle, P. F. Fracassi, R. Righini and S. Califano, Chem. Phys., 1983, 74, 179–195 CrossRef CAS.
- J. H. Roh, V. N. Novikov, R. B. Gregory, J. E. Curtis, Z. Chowdhuri and A. P. Sokolov, Phys. Rev. Lett., 2005, 95, 038101 CrossRef CAS PubMed.
- M. D. King, W. D. Buchanan and T. M. Korter, J. Phys. Chem. A, 2010, 114, 9570–9578 CrossRef CAS PubMed.
- T. M. Korter and D. F. Plusquellic, Chem. Phys. Lett., 2004, 385, 45–51 CrossRef CAS.
- V. Barone, M. Biczysko and J. Bloino, Phys. Chem. Chem. Phys., 2014, 16, 1759–1787 RSC.
- F. Pascale, C. M. Zicovich-Wilson, F. L. Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- C. M. Zicovich-Wilson, F. Pascale, C. Roetti, V. R. Saunders, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 1873–1881 CrossRef CAS PubMed.
- R. E. Allen and F. W. De Wette, Phys. Rev., 1969, 179, 873–886 CrossRef CAS.
- S. Baroni, P. Giannozzi and E. Isaev, Rev. Mineral. Geochem., 2010, 71, 39–57 CrossRef CAS.
- A. Erba, J. Maul and B. Civalleri, Chem. Commun., 2016, 52, 1820–1823 RSC.
- C. Cervinka, M. Fulem, R. P. Stoffel and R. Dronskowski, J. Phys. Chem. A, 2016, 120, 2022–2034 CrossRef CAS PubMed.
- Y. N. Heit and G. J. O. Beran, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 514–529 CAS.
- J. Hoja, A. M. Reilly and A. Tkatchenko, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1294 CrossRef.
- E. H. Linfield, Y. C. Shen, P. C. Upadhya and A. G. Davies, Appl. Phys. Lett., 2003, 82, 2350 CrossRef.
- M. T. Ruggiero and J. A. Zeitler, J. Phys. Chem. B, 2016, 120, 11733–11739 CrossRef CAS PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, Ph. D'Arco, Y. Noël, M. Causá, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Brandenburg and S. Grimme, Prediction and Calculation of Crystal Structures, Springer International Publishing, 2014, vol. 345, pp. 1–23 Search PubMed.
- L. Maschio, B. Kirtman, M. Rérat, R. Orlando and R. Dovesi, J. Chem. Phys., 2013, 139, 164101 CrossRef PubMed.
- L. Maschio, B. Kirtman, M. Rérat, R. Orlando and R. Dovesi, J. Chem. Phys., 2013, 139, 164102 CrossRef PubMed.
- L. Maschio, B. Kirtman, M. Rérat, R. Orlando and R. Dovesi, J. Chem. Phys., 2013, 139, 167101 CrossRef PubMed.
- A. Erba, J. Chem. Phys., 2014, 141, 124115 CrossRef CAS PubMed.
- A. Erba, M. Shahrokhi, R. Moradian and R. Dovesi, J. Chem. Phys., 2015, 142, 044114 CrossRef CAS PubMed.
- A. Erba, J. Maul, R. Demichelis and R. Dovesi, Phys. Chem. Chem. Phys., 2015, 17, 11670–11677 RSC.
- A. Erba, J. Maul, M. De La Pierre and R. Dovesi, J. Chem. Phys., 2015, 142, 204502 CrossRef CAS PubMed.
- A. Erba, J. Maul, M. Itou, R. Dovesi and Y. Sakurai, Phys. Rev. Lett., 2015, 115, 117402 CrossRef CAS PubMed.
- C. M. Foster, Z. Li, M. Grimsditch, S.-K. Chan and D. J. Lam, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10160–10167 CrossRef CAS.
- CCDC 1528479 contain supplemental crystallographic data for this paper.
- D. G. Watson, R. M. Sweet and R. E. Marsh, Acta Crystallogr., 1965, 19, 573–580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1528479. Additional data related to this publication are available at the University of Cambridge data repository (DOI: 10.17863/CAM.8309). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc00509a |
| This journal is © The Royal Society of Chemistry 2017 |