
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical [11C]CO2 to [11C]CO conversion for PET imaging†
David A.
Anders
ab,
Salvatore
Bongarzone
b,
Robin
Fortt
b,
Antony D.
Gee
*b and
Nicholas J.
Long
 *a
*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: n.long@imperial.ac.uk
bDivision of Imaging Sciences and Biomedical Engineering, King's College London, 4th Floor Lambeth Wing, London SE1 7EH, UK
First published on 24th February 2017
Abstract
The development of a novel electrochemical methodology to generate carbon-11 carbon monoxide ([11C]CO) from cyclotron-produced carbon-11 carbon dioxide ([11C]CO2) using Ni(cyclam) and Zn(cyclen) complexes is described. This methodology allows up to 10% yields of [11C]CO from [11C]CO2. Produced [11C]CO was subsequently converted to [11C]N-benzylbenzamide under mild conditions with a radiochemical purity (RCP) of >98%.
Electrochemical reduction of CO2 to CO has long been considered an important issue for tackling environmental sustainability. The challenge lies in converting the thermodynamically stable CO2 molecule into more energetic compounds. The largest thermodynamic barrier is the first electron addition to convert the linear CO2 molecule to a bent anion radical (COO˙) at E = −1.90 V.1 So far, 2nd and 3rd row transition metal elements have dominated this area although only Au and Ag generate CO with Faradaic efficiencies (FE) above 80% and maintain high current densities.2,3 Their activities have been boosted by nanostructuring techniques controlling the surface morphology.4 Other cheaper metals such as Sn, Sb, Pb and Bi have been used to convert CO2 to CO with high current efficiencies using ionic liquids to stabilise the COO˙ intermediate.5 Ionic liquids have been increasingly studied for their role in lowering thermodynamic barriers in CO2 reduction and so too have group 1 cations such as K+ and Cs+.6
Homogenous electrocatalysts for CO2 reduction, such as cathode materials, have previously been dominated by metals such as Pd, Ru, Rh and Re.7 Over the last few decades, many complexes of the first row transition metal elements such as Fe, Co, Cr, Cu, Mn and Ni have been used as electrocatalysts. Most notably metal cyclams (metal = Ni and Co),8 metalloporphyrins (Fe, Co and Ni),9 metal polypyridines (Cr, Fe, Co and Ni), and metal phthalocyanines (Ni, Co, Mn, Fe and Cu).10 Most of these catalysts act as electron shuttles between the electrode and the CO2 molecule and so generally the metal is in a low oxidation state and the ligand stabilises the intermediates by some inner-sphere effect. Recently, several groups have also highlighted that the activity of these catalysts can be boosted by adding protons on addition of mild acids (CF3CH2OH)11 and further increases in activity were realised when these acidic groups were added to the surrounding ligand.9
One of the most well-studied transition metal catalysts is Ni(cyclam)2+ which demonstrates very good CO selectivity at relatively low overpotentials in aqueous conditions. Most studies have been conducted at a Hg electrode due to the large negative potential window. Furthermore, Ni(cyclam)+ has been shown to adsorb to the Hg electrode and increase its reactivity to CO2 as a result.12 Recent studies by Kubiak and co-workers have demonstrated effective CO2 reduction at a glassy carbon electrode13 with the catalyst efficiency boosted by a CO scavenger [Ni(tetramethylcyclam)]2+.14
Our interest was to apply the electrochemical reduction to carbon-11 CO2 ([11C]CO2) generating [11C]CO. The range of functionalities that can be synthesised from [11C]CO make it an attractive precursor for positron emission tomography (PET) radiotracer development.15,16 However, the poor solubility of [11C]CO in organic solvents and low partial pressure, adds to the challenge of a short half-life (t1/2 = 20.4 min). A number of methodologies have been developed to convert cyclotron-produced [11C]CO2 to [11C]CO: (1) gas phase reduction method, which involves passing [11C]CO2 through a heated column of zinc or molybdenum at 400 °C or 850 °C respectively.17 Whilst molybdenum is preferred, both methods suffer reliability and repeatability issues making clinical production difficult from a regulatory stand-point; (2) chemical reduction methods that have been trialled use reactive silane lithium reagents that must be prepared beforehand.18
The aim of this work was to conduct a proof-of-principle study into the viability of electrochemical [11C]CO2 reduction to [11C]CO within a radiochemical setting. Trapping efficiencies represent decay-corrected trapped radioactivity as a percentage of dispensed radioactivity. RCY's are decay corrected and are estimated from dispensed [11C]CO2 converted to [11C]N-benzylbenzamide (5).
A DropSens® screen printed electrode with a carbon working electrode (WE), a counter electrode (CE) and a silver reference electrode (RE) was used for the electrochemical conversion (Fig. 1A). Fig. 1B and C show how the electrodes and electrode connector fit inside Vial A.
 | ||
| Fig. 1 (A) DropSens® screen printed electrode with carbon working electrode (WE), a counter electrode (CE) and a silver reference electrode (RE). (B) Screen printed electrode attached to cables via electrode connector (yellow rectangle) to the potentiostat through a silicone septum. (C) Electrode and connectors inserted in custom made screw-top vial. | ||
Initial experiments were conducted using a two vial set-up (set-up I – Vials A and B, Fig. 2). Vial A (used to convert [11C]CO2 to [11C]CO) contains the electrodes and the electrocatalysts Ni(cyclam)2+ or Zn(cyclen)2+ (1–2, Scheme 1) complexes in 0.1 M KCl(aq.) solution at 20 °C.19 Vial B (used to trap and fix [11C]CO) containing the carbonylation reagents to produce [11C]N-benzylbenzamide ([11C]5, Scheme 1).20 An ascarite trap was placed between the two vials to capture any untrapped [11C]CO2 (Fig. 2).
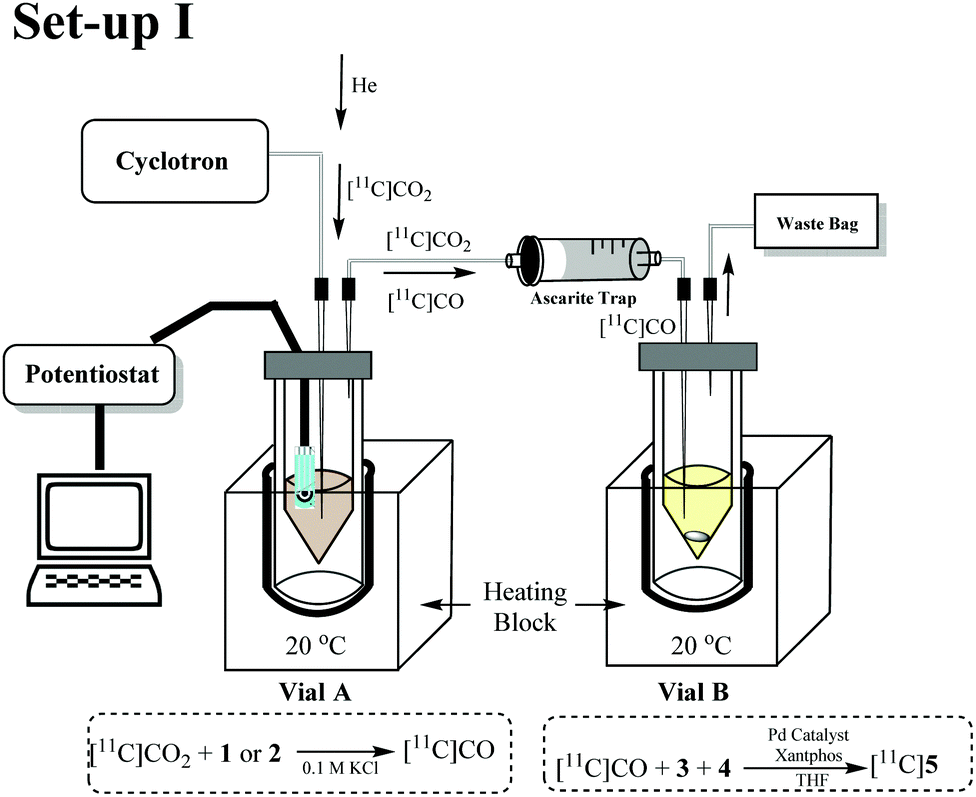 | ||
| Fig. 2 Schematic of the two-vial, one-valve setup. (1) [11C]CO2 delivered to Vial A with potentiostat switched on for 5 min (prior to and during delivery). (2) Helium sweep gas applied for 30 s through Vial B. (3) Carbonylation reaction in Vial B conducted for 10 min at 40 °C. | ||
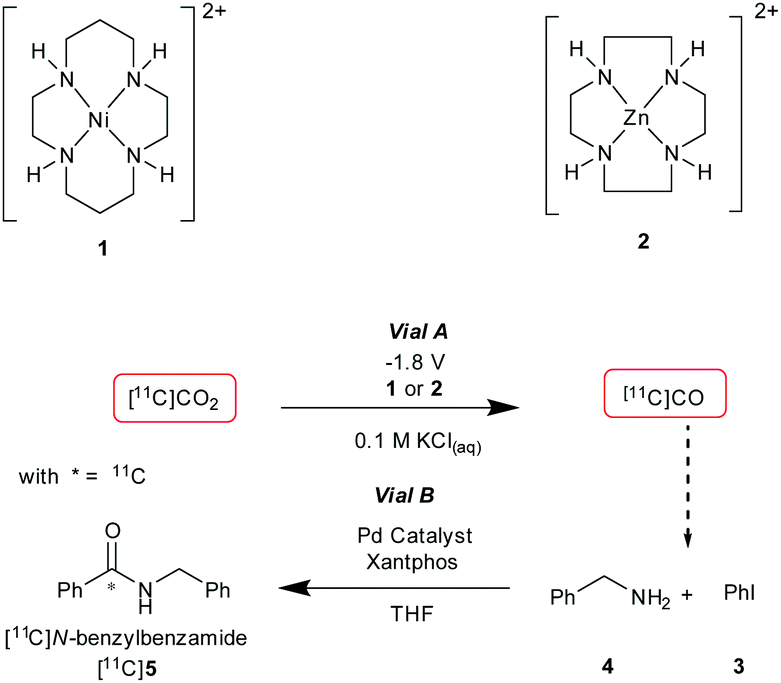 | ||
| Scheme 1 Vial A: catalyst 1 or 2, 0.1 M KCl, −1.8 V for 5 min. Vial B: [11C]CO, iodobenzene (3, 0.01 mmol), benzylamine (4, 0.46 mmol), [(Cinnamyl)PdCl]2 (0.007 mmol), Xantphos (0.007 mmol), THF (0.5 mL), 20 °C, 6 min. | ||
The setup tested is shown in Fig. 2. As the first experiment, [11C]CO2 was bubbled through the system with no potential applied to the electrodes. A low percentage (<1%) of [11C]5 (Table 1, entry 1) was detected and this was believed to be from cyclotron generated [11C]CO. When a potential of −1.8 V vs. Ag/AgCl was applied (in non-radioactive experiments (see ESI†) potentials of −1.4 and −1.6 V were used to allow full quantification of CO production, at −1.8 V, the detector was quickly saturated by CO. At more negative potentials H2 production was thought to become more favoured), [11C]5 was produced with high RCP's (>98%) but low RCY's (Table 1, entries 2 and 3). The low RCY was thought to be due to the low trapping efficiency of [11C]CO2 within Vial A. From these preliminary results it appeared that complex 2 performed marginally better than complex 1. The predicted trapping of [11C]CO as an adduct of 114 (ESI,† S2) was not observed in any usable quantity so experiments were conducted with 2.
| Entry | Complex | E app (V) | Radioactivity Vial A remaining at EOS (%) | Radioactivity Vial B at EOS (%) | RCP [11C]5a (%) | RCY [11C]5b (%) |
|---|---|---|---|---|---|---|
| Radioactivity distribution for reduction of [11C]CO2 to [11C]CO and subsequent [11C]CO capture conducted with 1 (50 mg, 227 mmol) or 2 (65 mg, 227 mmol) in 0.1 M KCl(aq.) (1 mL).a Radiochemical purity (RCP) of [11C]5 determined by analytical radio HPLC.b Decay corrected radiochemical yields (RCY) are based on the radioactivity of Vial B multiplied by the radiochemical purity of [11C]5 compared to the total radioactivity measured at end of cyclotron target bombardment (EOB). n = 2. End of synthesis (EOS). | ||||||
| 1 | 1 | 0 | 5.5 | 0.5 | 75 | <1 |
| 2 | 1 | −1.8 | 3.5 | 7.2 | 97 | 7 |
| 3 | 2 | −1.8 | 1.2 | 9.8 | 98 | 10 |
In order to evaluate and improve the trapping of [11C]CO2 in Vial A we designed a two-vial, one-valve set-up (set-up II) shown in Fig. 3. During [11C]CO2 delivery, Vial A was connected to ascarite 1 (Eckert & Ziegler Modular-Lab). By placing ascarite 1 after Vial A, the amount of [11C]CO2 trapped in Vial A before starting the electrolysis step could be assessed. At end of delivery (EOD) electrolysis would begin at −1.8 V and on completion of electrolysis, the valve was moved to divert gases ([11C]CO2 and [11C]CO) to Vial B by Helium purge. Ascarite 2 was placed after Vial B so that relative amounts of [11C]CO2 and [11C]CO (the latter assumed to be converted to [11C]5) in Vial B could be established by radio HPLC (see ESI†).
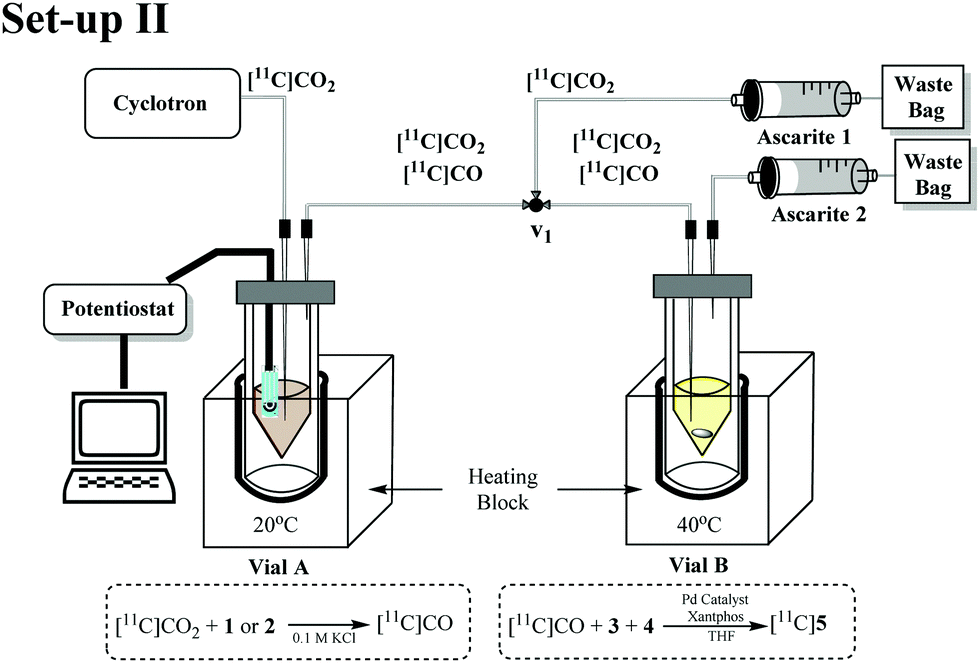 | ||
| Fig. 3 Schematic of the two-vial, one-valve setup. (1) [11C]CO2 delivered to Vial A. (2) potentiostat switched on for 10 min. (3) He sweep gas applied for 10 s through Vial B. (4) carbonylation reaction in Vial B conducted for 10 min at 40 °C. | ||
The performance of set-up II was evaluated using complex 2 at 150 and 15 mM (Table 2, entries 1 and 2). Increasing the concentration of 2 from 15 to 150 mM resulted in higher trapping (56% and 66% respectively) and conversion (<1% and 4% RCY, respectively). The improvements in trapping [11C]CO2 have been previously achieved using bases such as diazabicyclo[5.4.0]undecene (DBU), 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP), tetramethylethylenediamine (TMEDA)21 and triethanolamine (TEA).22 For our study we chose the strong base DBU and a weaker base TEA.
| Entry | 2 (mM) | Base (mM) | HCl added (0.1 N, 0.2 mL) | Est. [11C]CO2 trapping in Vial A at EODa (%) | RCPb [11C]5 (%) | RCY [11C]5c (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: 2 (15–150 mmol), base (7.5–75 mM) in 0.1 M KCl(aq.) (1 mL). The acid was added after peak [11C]CO2 trapping in Vial A was achieved.a Trapping in A = % radioactivity in Vial A versus total radioactivity released by the cyclotron.b RCP determined by analytical radio-HPLC.c Radiochemical yield (RCY) = [(radioactivity in Vial B × RCP [11C]5)/(radioactivity in Vial A at EOD) × 100].d Only WE and CE used. | ||||||
| 1 | 150 | — | — | 66 | 7 | 4 |
| 2 | 15 | — | — | 56 | 3 | 1 |
| 3 | 150 | DBU (75) | — | 80 | — | — |
| 4 | 150 | DBU (75) | ✓ | 48 | 3 | 3 |
| 5 | 150 | DBU (7.5) | ✓ | 18 | — | — |
| 6 | 150 | TEA (75) | — | 23 | — | — |
| 7 | 150 | TEA (75) | ✓ | 65 | 29 | 1 |
| 8 | 150 | TEA (7.5) | ✓ | 60 | 7 | 5 |
| 9 | 15 | TEA (75) | ✓ | 32 | 10 | <1 |
| 10 | 15 | TEA (7.5) | ✓ | 35 | 8 | 2 |
| 11d | 150 | — | — | 12 | 8 | 3 |
| 12d | 150 | TEA (75) | ✓ | 64 | 5 | <1 |
| 13d | 15 | TEA (7.5) | ✓ | 52 | 20 | 6 |
The next experiments were performed using 150 mM of 2. To improve [11C]CO2 trapping, DBU was added. A high concentration of base was used initially to improve the trapping of [11C]CO2 and subsequently promote the formation of [11C]5. Although 75 mM of bases increased the trapping of [11C]CO2 (Table 2, entry 1 versus entry 3) the high concentration appeared to prevent the formation of [11C]CO. No conversion was observed until HCl was added (0.2 mL of 0.1 N HCl, Table 2, entry 4). This was thought to be due to a change of pH from 6.5 to 12–13 from using DBU in water solution. The resultant acidification of Vial A (on addition of HCl) released the trapped [11C]CO2 so it was free to be converted to [11C]CO.
On addition of HCl, entry 4 showed a RCY of 3%. A lower concentration of base (7.5 mM) was used to investigate if the concentration of 2 and the concentration of base had an optimum combination, perhaps acting through the base binding with 2. Substitution of DBU for TEA was added as a more moderate base (pH 9–10) and the trapping efficiency varying from 23–65% in Vial A (Table 2, entries 6–10). Table 2, entries 7 and 8, showed trapping efficiencies of ∼60%. On reducing the concentration of 2 to 15 mM, the trapping efficiency is halved (∼30%) irrespective of the concentration of TEA suggesting that the concentration of 2 plays a larger role than TEA in trapping [11C]CO2. This variety of trapping efficiencies shown in Table 2 was thought to be a consequence of the high flow rate (50 ml min−1) of [11C]CO2 into an aqueous solution.23
The optimum RCY achieved (Set-up II, Fig. 3) was 5% (Table 2, entry 8) which was obtained when 7.5 mM of TEA was used. These results appeared to show that a compromise of a milder base would still facilitate reasonable trapping whilst not hindering [11C]CO2 reduction. In order to simplify the reaction set-up and increase electrode surface area, a 2-electrode set-up was used in Vial A. This involved using just the working electrode (WE) and the counter electrode (CE) (Table 2, entries 11–13). The optimum conversion achieved by the 2-electrode cell was 6% (Table 2, entry 13) with 52% of [11C]CO2 initially trapped in Vial A.
In conclusion, the first electrochemical [11C]CO2 to [11C]CO reduction has been achieved with a 2-vial set-up to incorporate the [11C]CO product into [11C]N-benzylbenzamide in a proof-of-principle study. 2 showed good [11C]CO2 trapping and conversion to [11C]CO. The effectiveness of 2 compared to 1 for [11C]CO2 reduction was surprising and further studies are needed to investigate this fully although we believe that ZnO nanoparticles are being generated at the electrode. Improvements in the performance of 1 could come from binding the catalyst to the electrode.8,25 Furthermore, the application of a two-electrode design of Vial A was shown to be viable. We believe that a pre-concentration step of [11C]CO2 prior to vial A would lead to better performance both in trapping of [11C]CO2 and conversion.
This work was supported by the Medical Research Council through financial support (MRC-1527506 and MR/K022733/1) and the Institute of Chemical Biology at Imperial College London. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust and the Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC under grant number WT 088641/Z/09/Z.
Notes and references
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036 RSC.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1987, 728 RSC.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969 CrossRef CAS PubMed; A. Salehi-Khojin, H.-R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627 Search PubMed; Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 4242 Search PubMed.
- J. Medina-Ramos, R. C. Pupillo, T. P. Keane, J. L. Dimeglio and J. Rosenthal, J. Am. Chem. Soc., 2015, 137, 5021 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328 RSC; J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Organometallics, 2006, 25, 3345 CrossRef CAS; K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef; S. Slater and J. H. Wagenknecht, J. Am. Chem. Soc., 1984, 106, 5367 CrossRef; J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283 CrossRef PubMed.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461 CrossRef CAS PubMed; J. L. Karn and D. H. Busch, Inorg. Chem., 1969, 8, 1149 CrossRef; G. Neri, J. J. Walsh, C. Wilson, A. Reynal, J. Y. C. Lim, X. Li, A. J. P. White, N. J. Long, J. R. Durrant and A. J. Cowan, Phys. Chem. Chem. Phys., 2015, 17, 1562 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423 RSC; C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90 CrossRef CAS PubMed; C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990 CrossRef PubMed.
- J. L. Inglis, B. J. MacLean, M. T. Pryce and J. G. Vos, Coord. Chem. Rev., 2012, 256, 2571 CrossRef CAS.
- I. Bhugun, D. Lexa and J. Savéant, J. Am. Chem. Soc., 1996, 118, 1769 CrossRef CAS; J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484 CrossRef PubMed.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1993, 361, 149 CrossRef CAS; M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315 RSC.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932 CrossRef CAS PubMed.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565 CrossRef CAS PubMed.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed.
- S. Kealey, A. Gee and P. W. Miller, J. Labelled Compd. Radiopharm., 2014, 57, 195 CrossRef CAS PubMed; P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef PubMed.
- K. Dahl, O. Itsenko, O. Rahman, J. Ulin, C.-O. Sjöberg, P. Sandblom, L.-A. Larsson, M. Schou and C. Halldin, J. Labelled Compd. Radiopharm., 2015, 58, 220 CrossRef CAS PubMed; J. Eriksson, J. Hoek and A. D. Windhorst, J. Labelled Compd. Radiopharm., 2012, 55, 223 CrossRef; E. D. Hostetler and H. D. Burns, Nucl. Med. Biol., 2002, 29, 845 CrossRef PubMed.
- C. Taddei, S. Bongarzone, A. K. Haji Dheere and A. D. Gee, Chem. Commun., 2015, 51, 11795 RSC; P. Nordeman, S. D. Friis, T. L. Andersen, H. Audrain, M. Larhed, T. Skrydstrup and G. Antoni, Chem. – Eur. J., 2015, 21, 17601 CrossRef CAS PubMed.
- The use of Zn(cyclen) was as a result of studies that suggested favourable [11C]CO2 binding could be expected.24 Experiments conducted in a non-radiochemical setting showed comparable CO2 formation to that of the well-studied Ni(cyclam) complex. D. A. Anders, PhD thesis, Imperial College London, 2016, see ESI†.
- K. Dahl, M. Schou, N. Amini and C. Halldin, Eur. J. Org. Chem., 2013, 1228 CrossRef CAS.
- P. J. Riss, S. Lu, S. Telu, F. I. Aigbirhio and V. W. Pike, Angew. Chem., Int. Ed., 2012, 51, 2698 CrossRef CAS PubMed; J. M. Hooker, A. T. Reibel, S. M. Hill, M. J. Schueller and J. S. Fowler, Angew. Chem., Int. Ed., 2009, 48, 3482 CrossRef PubMed; A. A. Wilson, A. Garcia, S. Houle and N. Vasdev, Org. Biomol. Chem., 2010, 8, 428 Search PubMed.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825 CrossRef CAS PubMed.
- N. T. Vandehey and J. P. O’Neil, Appl. Radiat. Isot., 2014, 90, 74 CrossRef CAS PubMed.
- L. Koziol, C. A. Valdez, S. E. Baker, E. Y. Lau, W. C. Floyd, S. E. Wong, J. H. Satcher, F. C. Lightstone and R. D. Aines, Inorg. Chem., 2012, 51, 6803 CrossRef CAS PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc00319f |
| This journal is © The Royal Society of Chemistry 2017 |