
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted tumor detection: guidelines for developing biotinylated diagnostics†
Joo Hee
Jang‡
a,
Woo Ri
Kim‡
b,
Amit
Sharma‡
a,
Suk Hee
Cho
b,
Tony D.
James
 *c,
Chulhun
Kang
*b and
Jong Seung
Kim
*a
*c,
Chulhun
Kang
*b and
Jong Seung
Kim
*a
aDepartment of Chemistry, Korea University, Seoul, 136-701, Korea. E-mail: jongskim@korea.ac.kr
bThe School of East-West Medical Science, Kyung Hee University, Yongin, 446-701, Korea. E-mail: kangch@khu.ac.kr
cDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: T.D.James@bath.ac.uk
First published on 25th January 2017
Abstract
The challenge in achieving precision medicine relies on how to advance and/or enhance new as well as old therapeutic strategies. Here, we highlight the significant role hydrophilicity of biotinylated fluorescent probe's plays on their cellular uptake behaviour.
Cancer is a near-term objective of the precision medicine initiative (2015) which aims to revolutionize current health programs by tailoring therapeutics towards individual patients.1 The overarching goal behind this initiative is for medicinal care providers to decrease the cancer modalities and morbidities, regardless of the cancer subtype, contingent on the results obtained from reliable assays for relevant markers from patients, that are pertinent for disease prevention.2 However, this monumental task presents numerous biological challenges such as pathological and physiological complications, non-targeted delivery due to genetic heterogeneity within tumors, dynamic drug resistance, problems with cancer subtype classification and associated off-target side effects. With the revolution in cancer genomics, immense effort has been directed towards the development of non-invasive biomarkers for assessing and transferring genetic information into clinical practices in order to facilitate diagnosis, monitor the therapeutic response and patient stratifications.3 Generally, development programs are endorsed under regulatory guidelines for better outcomes from various clinical processes and implementations.4 However, regardless of these huge endeavors and resources provided, the real hurdles confronting the clinical development and endorsement of current precision based regimens are selectivity, tissue penetration with adequate uptake followed by pathways for drug clearing. In order to take full advantage of advanced cancer genomics, we require a better understanding of the mechanisms responsible for better targeted diagnostics leading to improved therapeutics.
Biotin (vitamin H), a critical cofactor for carboxylase activity, is involved in fatty acid synthesis, branched amino acids catabolism and gluconeogenesis5,6 and is preferentially delivered into rapidly proliferating cells including cancer cells (ovarian, colorectral, etc.), through the overexpressed sodium dependent multivitamin transporter (SMVT) on the cell surface, whose activity is additionally regulated by protein kinase C (PKC).6,7 Biotin-conjugation is one of the most plausible choices in developing various cancer selective prodrugs, polymeric carriers for drug delivery, and theranostic systems.8,9 Recently, biotin has been extensively used as a protein labeling tool in order to identify various protein interactions.10 In theranostic biotin-conjugates, a biotin unit is linked through a self-immolative linker to the drug molecule, which may vary from lipophilic drug11 to highly polar peptide.12 In general, cellular uptake of hydrophilic molecules may be aided by the corresponding membrane proteins, whereas the hydrophobic molecules, for instance, steroid hormones, simply diffuse through the membrane.13
In this context, despite the incorporation of a biotin moiety for SMVT targeting, one may ask whether or not the hydrophobicity of biotin-conjugates plays an adverse role in their cellular uptake. Moreover, the biological pathway responsible for theranostic biotin-conjugate's uptake still remains elusive. These factors are critical for the development of smarter biotin-conjugates in order to widen the tenets of precision medicine with an emphasis on disease prevention.
In order to investigate the role of hydrophilicity in biotinylated conjugate towards cellular uptake, we designed biotin-conjugated fluorescent probes (4–6) (Scheme 1), possessing dodecyl, hexyl, diethylene glycol to adjust their overall hydrophobicity and the corresponding non-biotin analogs (1–3) were used as controls (Scheme 1). The 4-amino-1,8-naphthalimide fluorophore was chosen due to its strong emission. Additionally, the two photon properties can afford better cell imaging efficiency with minimal background and enhanced light penetration. The in vitro cellular uptake behavior and mechanism were investigated against HeLa cells. Compared to other conjugates, biotin fluorescent probe 5 with log Poct ∼ 1 exhibited preferential cell membrane uptake via SMVT proteins under PKC-mediation using intracellular ATPs.
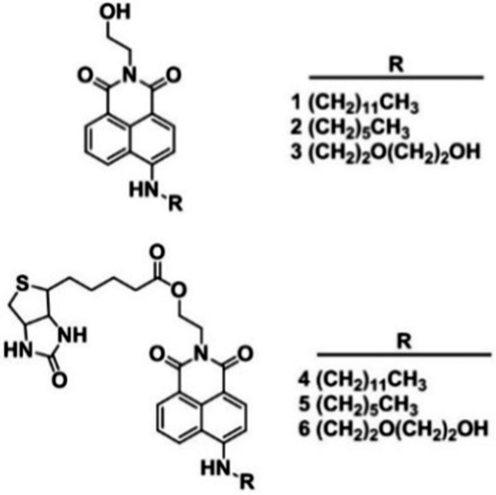 | ||
| Scheme 1 Chemical structure of the non-biotin (1–3) and biotin-conjugated probes (4–6). | ||
Compounds 1–6 were synthesized according to Scheme S1 (ESI†) in good yield. Spectroscopic studies of the probes were undertaken using UV/Vis absorption and fluorescence spectroscopy. These probes exhibit a broad absorption band at 430 (in toluene), 433 (in acetonitrile), and 440 nm (in PBS) respectively, (Fig. S1, ESI†) with a bandwidth of ∼75 nm. The corresponding fluorescence emission bands of the probes in toluene and acetonitrile were slightly blue-shifted with enhanced intensities (505 and 524 nm respectively) when compared to the PBS (545 nm) system. Since the internal charge-transfer (ICT) excited state phenomenon in these probes offers a substantial excited-state dipole moment, which can be stabilized/destabilized depending upon the choice of solvent. The lower fluorescent emission intensity of the probes in PBS is ascribed to their hydrogen bond donor (HBD) or hydrogen bond acceptor (HBA) ability.14
To examine the cell uptake behavior of biotin-conjugates, we performed fluorescent confocal microscopic experiments in HeLa cells for each conjugate (1–6). As shown in Fig. S2a (ESI†), probe 1 with a dodecyl group displayed the strongest fluorescence intensity among the non-biotin probes (1–3) and its intensity is expected to keep on increasing even after 60 min. Due to the more hydrophobic tail of this molecule, it penetrates into the cell membrane via diffusion. Conversely, among the biotin-conjugates (4–6), the largest intensity was observed for the system with a hexyl moiety (5), while one with a more hydrophobic dodecyl (4) and a hydrophilic hydroxylethyl–oxyethyl moiety (6) do not display comparable fluorescence intensity (Fig. S2b, ESI†). These results indicate that the biotin-conjugates enter the cells through an alternate pathway from that of the non-biotin counterparts.
In order to investigate the detailed cellular uptake mechanism of the biotin-conjugates (4–6), a time course analysis of the fluorescence intensity in the cells was carried out for probes 2 and 5 at various concentrations. We have found that the enhancement of fluorescence displays both time and concentration dependent behavior (Fig. 1). And, probe 5 shows 6 times higher fluorescence intensity than 2 incorporating the same alkyl chain, indicating that the biotin moiety plays a significant role in the cellular uptake. However, what makes the dramatic difference of uptake behavior among the biotin-conjugates shown in Fig. S2b (ESI†). The most apparent difference among the conjugate structures was the probes' hydrophilicity. Thereby, we measured the partition-coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Poct) of the probes according to the solubility in n-octanol and MOPS buffer (Table S1, ESI† and Fig. 2). The logPoct values for 4–6 are 1.29, 1.03, and 0.807, respectively. These results suggest an optimum hydrophobicity for the biotin-conjugates with a logPoct value of about 1.0.
Poct) of the probes according to the solubility in n-octanol and MOPS buffer (Table S1, ESI† and Fig. 2). The logPoct values for 4–6 are 1.29, 1.03, and 0.807, respectively. These results suggest an optimum hydrophobicity for the biotin-conjugates with a logPoct value of about 1.0.
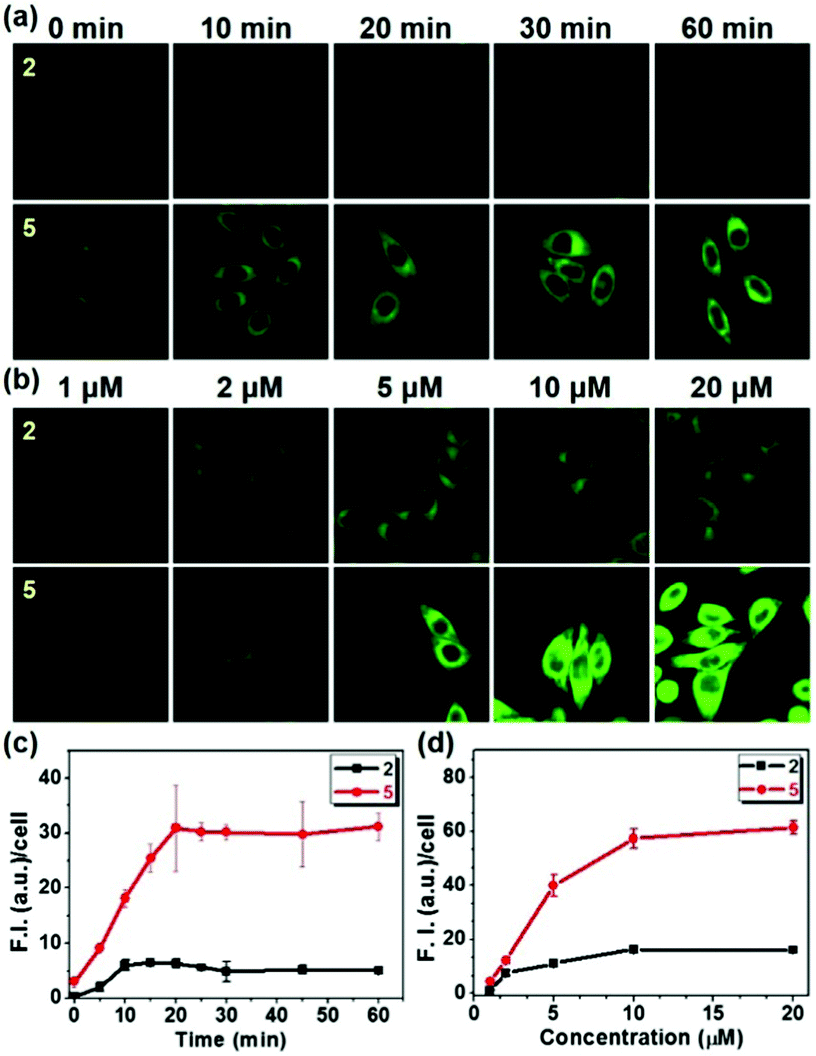 | ||
| Fig. 1 Confocal microscopy images of HeLa cells upon treatment with probes 2 and 5. (a) The fluorescent images were obtained at variable time (2 μM each); (b) and at variable probe's concentration for 10 min. The cells were incubated in high glucose serum free DMEM media at 37 °C, λex = 458 nm, bandpath filter (505–550 nm). (c and d) Fluorescence intensities (a.u.) per cell in the images of the panel (a) and (b) respectively. The images were obtained using Image J software. The data are presented as mean ± SD (n = 5). | ||
 | ||
| Fig. 2 Lipophilicity of probes (1–6) according to logPoct value. | ||
Subsequently, in order to identify the intracellular location of 2 and 5, a series of colocalization experiments were performed using commercially available organelle selective markers. As seen in Fig. S3 (ESI†), the green-channel fluorescence intensity of 5 showed an excellent overlay with the red-channel fluorescence of ER (Pearson's correlation coefficient (PCC) is 0.9729), whereas the overlap with other trackers were relatively poor (Lyso; 0.4617 and Mito; 0.5576). Its preference for the ER could be explained by the fact that the ER is the metabolic center of various lipophilic compounds such as xenobitics and lipids with the largest membrane area among the organelles. In contrast, the non-biotin-conjugate, 2 was localized in the ER as well as mitochondria (PCC = 0.9373 and 0.7809, respectively).
It is well known that biotin's uptake is regulated through SMVT, and it would be natural to ask whether biotin-conjugates enter the cells via the same mechanism. To the best of our knowledge, this has never been reported before. The uptake of probe 5 in Fig. 1 was examined in the presence of biotin and pantothenic acid, a well-known substrate for SMVT, and in the presence of folic acid and ascorbic acid for comparison (Fig. 3). Biotin and pantothenic acid induced significant inhibitory effects on the uptake of 5 in a concentration dependent manner whereas ascorbic acid and folic acid display no such effect. Similarly, a series of parallel experiments were also performed for the uptake of probe 2, the results clearly indicate that all vitamins used in this study fail to induce any inhibitory effect on the uptake of probe 2 (Fig. S4, ESI†). These results indicate that 5 enters the cells selectively via SMVT, whereas the transport of 2 is independent of SMVT, confirming the differential uptake processes.
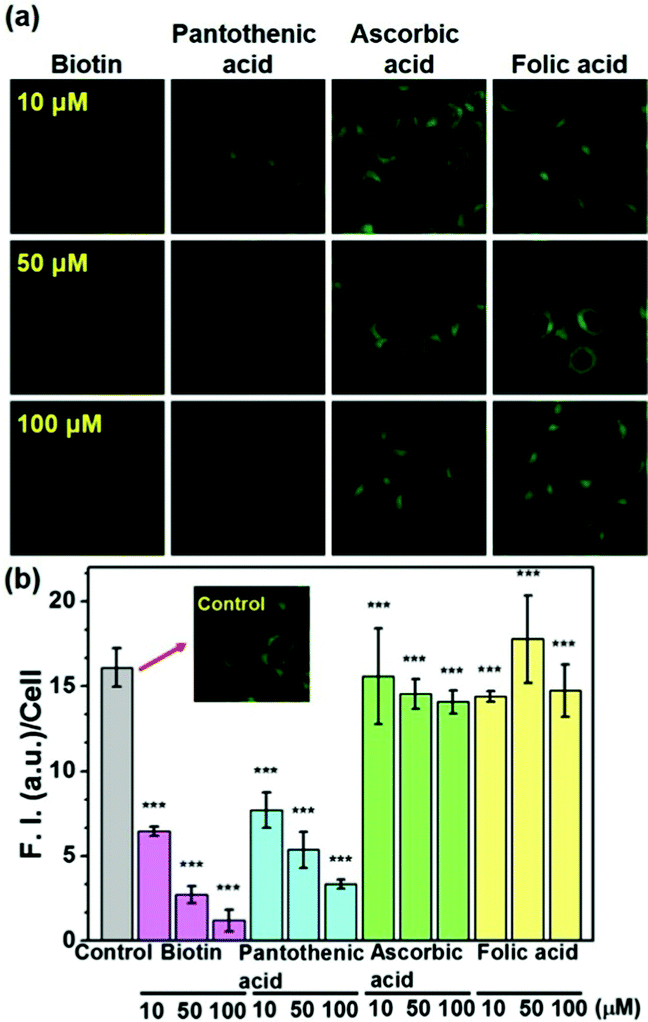 | ||
| Fig. 3 (a) Effect on the uptake of 5 in HeLa cells in the presence of vitamins biotin, pantothenic acid, ascorbic acid and folic acid. The cells were incubated with 2 μM of the probe in presence of the vitamin under high glucose serum free DMEM media at 37 °C, λex = 458 nm, bandpath filter (505–550 nm). (b) Fluorescence intensity (F.I.) per cell in the images of the panel (a). The images were analysed by Image J software. The data are presented as mean ± SD (n =5). | ||
Next, to demonstrate sodium ion-dependency for the uptake of 5, its cellular uptake behavior was examined in the presence of sodium ions and selective inhibitors for the sodium pump. As shown in Fig. S5 (ESI†), depletion of sodium ions by replacing the salts with potassium salts in the media induced a drastic decrease in the uptake of 5. This result indicates that the sodium gradient across the plasma membrane is the driving force of the biotin-conjugate's uptake, which is further confirmed by the inhibition of uptake by amiloride, a sodium ion transport inhibitor15 (Fig. S5a and b, ESI†). The replacement of chloride to phosphate in the media did not show any significant change in the uptake of probe 5. A gradual increase in 5's cellular uptake is observed according to the increase of sodium ions in the range 0–100 mM (Fig. S5c, ESI†). Similar experiments for the corresponding non-biotin probe 2 did not show any significant change (Fig. S5a, b, and S6, ESI†).
Considering that the sodium-dependent nutrient uptake as shown in this study is a typical characteristic of the secondary pump, biotin-conjugate uptake will depend on the cellular ATP level, which is examined in Fig. S7 (ESI†). Indeed, the uptake is clearly reduced by the presence of ouabain,16 sodium azide17 or 2,4-dinitrophenol,18 which are an Na+/K+-ATPase inhibitor, an oxidative phosphorylation inhibitor and a mitochondrial uncoupler, respectively and where all the chemicals reduce the cellular ATP level. Taken together with the results from Fig. 3 and Fig. S5 (ESI†), the results in Fig. S7 (ESI†) indicate that the cellular uptake of the biotin-conjugate in this study is via SMVT, a secondary pump whose driving force is regulated by intracellular ATP hydrolysis.
To check whether or not the physiological regulation of the SMVT activity for biotin uptake into cells works for the biotin-conjugates, the uptake of 5 into cells was investigated in the presence of the modulators for various signaling pathways; genistein,19 forskolin,20 PMA21 and KN-6222 are used to modulate the protein tyrosine kinase (PTK), protein kinase A (PKA) as well as C (PKC) and calcium–calmodulin pathways, respectively. As shown in Fig. 4, the fluorescence intensity from 5 is strongly inhibited by PMA, whereas the others did not show any effect, indicating that the biotin-conjugates uptake is selectively controlled by protein kinase C activity. These results also demonstrate that the biotin-conjugates are excellent candidates for cancer targeting imaging and drug delivery. Conversely, the uptake of the non-biotin system 2 was not affected by the presence of any of these modulators (Fig. S8, ESI†). Additionally, the viability of the cells was not affected by the presence of the biotin conjugates (Fig. S9, ESI†).
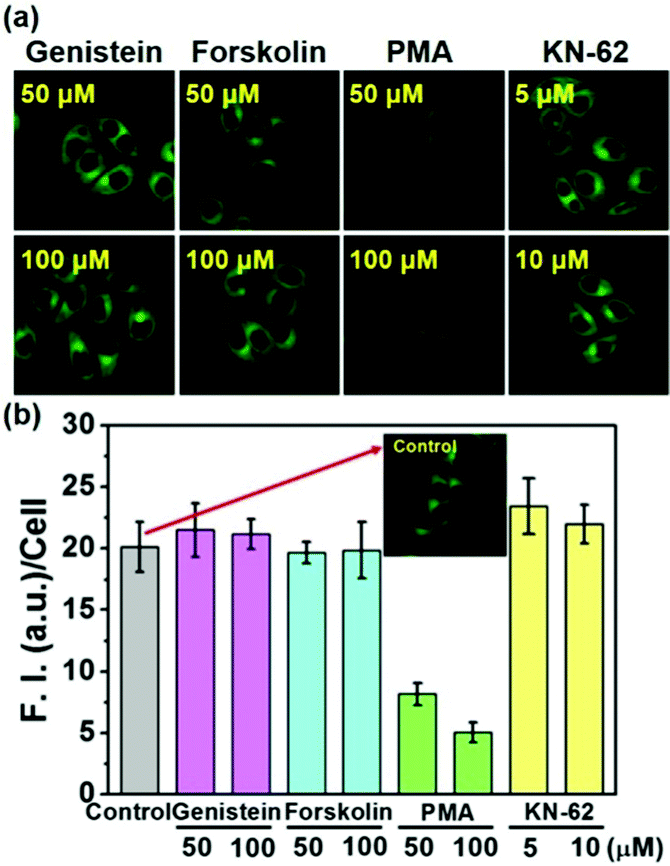 | ||
| Fig. 4 Effect of various signal transduction pathway modulators on the uptake of 5 in HeLa cells such as genistein, forskolin, PMA and KN-62. (a) After preincubation of the cells with inhibitors for 1 h. the cells were further incubated with 2 μM of a probe for 10 min. Under high glucose serum free DMEM media at 37 °C. λex = 458 nm, bandpath filter (505–550 nm). (b) Fluorescence intensity (F.I.) per cell in the images of the panel (a). The images were obtained using Image J software. The data are presented as mean ± SD (n =5). | ||
In summary, we have designed and developed biotinylated probes with varying hydrophilicity to investigate their cellular uptake mechanism and behavior. Compared with the non-biotinylated fluorescent probes, probe 5 exhibits preferential cellular uptake among other biotinylated probes through SMVT-protein receptors under sodium-ion dependent manner. In addition, the cellular uptake behavior is regulated under PKC-mediation by utilizing intracellular ATPs. Taken together, these data collectively highlight the critical role of hydrophilicity on the cellular uptake processes of biotin-based cancer targeting imaging agents. The use of these guidelines will expand the current cancer cell labelling/targeting toolbox and also offer the potential to improve their cellular uptake, which is crucial for the development of in vivo imaging systems and facilitate rational screening and allow for efficient diagnosis and monitoring of treatment response and more importantly patient satisfaction allowing for the implementation of precision medicine as part of standard patient care.
This work was supported by CRI (No. 2009-0081566, JSK) and NRF (No. 2015R1A5A1037656, CK) of Korea.
References
- (a) F. S. Collins and H. Varmus, N. Engl. J. Med., 2015, 372, 793 CrossRef CAS PubMed; (b) S. Hawgood, I. G. Hook-Barnard, T. C. O'Brien and K. R. Yamamoto, Sci. Transl. Med., 2015, 7, 300ps17 CrossRef PubMed; (c) L. A. Chantrill, A. M. Nagrial, C. Watson, A. L. Johns, M. Martyn-Smith, S. Simpson, S. Mead, M. D. Jones, J. S. Samra, A. J. Gill, N. Watson, V. T. Chin, J. L. Humphris, A. Chou, B. Brown, A. Morey, M. Pajic, S. M. Grimmond, D. K. Chang, D. Thomas, L. Sebastian, K. Sjoquist, S. Yip, N. Pavlakis, R. Asghari, S. Harvey, P. Grimison, J. Simes and A. V. Biankin, Clin. Cancer Res., 2015, 21, 2029–2037 CrossRef CAS PubMed.
- (a) A. Monica, V. Cecile, L. Sherene, L. Celine, M. Stefan, B. Herve and A. Fabrice, Nat. Rev. Clin. Oncol., 2015, 12, 693–704 CrossRef PubMed; (b) A. L. Richer, J. M. Friel, V. M. Carson, L. J. Inge and T. G. Whitsett, Pharmgenomics Pers. Med., 2015, 8, 63–79 CAS.
- (a) Y.-W. Jun, Y.-M. Huh, J.-S. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin, J.-S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed; (b) N. Kosaka, H. Iguchi and T. Ochiya, Cancer Sci., 2010, 101, 2087–2092 CrossRef CAS PubMed; (c) S. Ramaswamy, P. Tamayo, R. Rifkin, S. Mukherjee, C.-H. Yeang, M. Angelo, C. Ladd, M. Reich, E. Latulippe, J. P. Mesirov, T. Poggio, W. Gerald, M. Loda, E. S. Lander and T. R. Golub, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 15149–15154 CrossRef CAS PubMed.
- (a) C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS; (b) R. C. Bast, T. L. Klug, E. S. John, E. Jenison, J. M. Niloff, H. Lazarus, R. S. Berkowitz, T. Leavitt, C. T. Griffiths, L. Parker, V. R. Zurawski and R. C. Knapp, N. Engl. J. Med., 1983, 309, 883–887 CrossRef PubMed; (c) W. C. S. Cho, T. T. C. Yip, C. Yip, V. Yip, V. Thulasiraman, R. K. C. Ngan, T.-T. Yip, W.-H. Lau, J. S. K. Au, S. C. K. Law, W.-W. Cheng, V. W. S. Ma and C. K. P. Lim, Clin. Cancer Res., 2004, 10, 43–52 CrossRef CAS PubMed.
- (a) W. Yang, Y. Cheng, T. Xu, X. Wang and L. Wen, Eur. J. Med. Chem., 2009, 44, 862–868 CrossRef CAS PubMed; (b) J. Zempleni, S. S. Wijeratne and Y. I. Hassan, BioFactors, 2009, 35, 36–46 CrossRef PubMed.
- H. M. Said, J. Nutr., 1999, 129, 490S–493S CAS.
- (a) V. Kansara, S. Luo, B. Balasubrahmanyam, D. Pal and A. K. Mitra, Int. J. Pharm., 2006, 312, 43–52 CrossRef CAS PubMed; (b) H. M. Said, Subcell. Biochem., 2012, 56, 1 CAS.
- (a) S. W. Park, D. E. Casalena, D. J. Wilson, R. Dai, P. P. Nag, F. Liu, J. P. Boyce, J. A. Bittker, S. L. Schreiber, B. C. Finzel, D. Schnappinger and C. C. Aldrich, Chem. Biol., 2015, 22, 76–86 CrossRef CAS PubMed; (b) R. Kumar, J. Han, H.-J. Lim, W. X. Ren, J.-Y. Lim, J.-H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836–17843 CrossRef CAS PubMed; (c) M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed; (d) R. Kumar, E.-J. Kim, J. Han, H. Lee, W. S. Shin, H. M. Kim, S. Bhuniya, J. S. Kim and K. S. Hong, Biomaterials, 2016, 104, 119–128 CrossRef CAS PubMed; (e) W. S. Shin, J. Han, R. Kumar, G. G. Lee, J. L. Sessler, J.-H. Kim and J. S. Kim, Sci. Rep., 2016, 6, 29018 CrossRef CAS PubMed.
- (a) J. I. Stuckey, B. M. Dickson, N. Cheng, Y. Liu, J. L. Norris, S. H. Cholensky, W. Tempel, S. Qin, K. G. Huber, C. Sagum, K. Black, F. Li, X.-P. Huang, B. L. Roth, B. M. Baughman, G. Senisterra, S. G. Pattenden, M. Vedadi, P. J. Brown, M. T. Bedford, J. Min, C. H. Arrowsmith, L. I. James and S. V. Frye, Nat. Chem. Biol., 2016, 12, 180–187 CrossRef CAS PubMed; (b) J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850–11853 CrossRef CAS PubMed.
- (a) J. Wang, S. Rao, J. Chu, X. Shen, D. N. Levasseur, T. W. Theunissen and S. H. Orkin, Nature, 2006, 444, 364–368 CrossRef CAS PubMed; (b) H. Zhu, M. Bilgin, R. Bangham, D. Hall, A. Casamayor, P. Bertone, N. Lan, R. Jansen, S. Bidlingmaier, T. Houfek, T. Mitchell, P. Miller, R. A. Dean, M. Gerstein and M. Snyder, Science, 2001, 293, 2101–2105 CrossRef CAS PubMed.
- (a) S. R. Sirsi, C. Fung, S. Garg, M. Y. Tianning, P. A. Mountford and M. A. Borden, Theranostics, 2013, 3, 409–419 CrossRef CAS PubMed; (b) X. Liu, B. Testa and A. Fahr, Pharm. Res., 2011, 28, 962–977 CrossRef CAS PubMed.
- J. B. Rothbard, S. Garlington, Q. Lin, T. Kirschberg, E. Kreider, P. L. McGrane, P. A. Wender and P. A. Khavari, Nat. Med., 2000, 6, 1253–1257 CrossRef CAS PubMed.
- J.-P. Gratton, J. Yu, J. W. Griffith, R. W. Babbitt, R. S. Scotland, R. Hickey, F. J. Giordano and W. C. Sessa, Nat. Med., 2003, 9, 357–362 CrossRef CAS PubMed.
- P. Kucheryavy, G. Li, S. Vyas, C. Hadad and K. D. Glusac, J. Phys. Chem. A, 2009, 113, 6453–6461 CrossRef CAS PubMed.
- J. L. Kinsella and P. S. Aronson, Am. J. Physiol., 1981, 241, F374–F379 CAS.
- F. Proverbio, J. W. L. Robinson and G. Whittembury, Biochim. Biophys. Acta, Biomembr., 1970, 211, 327–336 CrossRef CAS.
- D. E. Keilin and E. F. Hartree, Proc. R. Soc. London, Ser. B, 1939, 127, 167–191 CrossRef CAS.
- (a) W. F. Loomis and F. Lipmann, J. Biol. Chem., 1948, 173, 807–808 CAS; (b) V. P. Skulachev, Biochim. Biophys. Acta, 1998, 1363, 100–124 CrossRef CAS PubMed.
- T. Akiyama, J. Ishida, S. Nakagawa, H. Ogawara, S. Watanabe, N. Itoh, M. Shibuya and Y. Fukami, J. Biol. Chem., 1987, 262, 5592–5595 CAS.
- K. B. Seamon and J. W. Daly, J. Cyclic Nucleotide Res., 1981, 7, 201–224 CAS.
- T. J. Rink, A. Sanchez and T. J. Hallam, Nature, 1983, 305, 317–319 CrossRef CAS PubMed.
- H. Tokumitsu, T. Chijiwa, M. Hagiwara, A. Mizutani, M. Terasawa and H. Hidaka, J. Biol. Chem., 1990, 265, 4315–4320 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc00311k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |