
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-selective direct 17O DNP NMR of CeO2 nanoparticles†
Michael A.
Hope
 a,
David M.
Halat
a,
Pieter C. M. M.
Magusin
a,
Subhradip
Paul
b,
Luming
Peng
c and
Clare P.
Grey
*a
a,
David M.
Halat
a,
Pieter C. M. M.
Magusin
a,
Subhradip
Paul
b,
Luming
Peng
c and
Clare P.
Grey
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: cpg27@cam.ac.uk
bDNP MAS NMR Facility, Sir Peter Mansfield Magnetic Resonance Centre, University of Nottingham, Nottingham, NG7 2RD, UK
cKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, China
First published on 24th January 2017
Abstract
Surface-selective direct 17O DNP has been demonstrated for the first time on CeO2 nanoparticles, for which the first three layers can be distinguished with high selectivity. Polarisation build-up curves show that the polarisation of the (sub-)surface sites builds up faster than the bulk, accounting for the remarkable surface selectivity.
Nanoparticulate transition metal oxides are of technological importance in various areas of chemistry and materials science, such as catalysis, energy storage and electronics.1–4 However, optimisation of materials for these applications necessitates thorough knowledge of structure–function relationships, which in turn requires an accurate description of the local surface structure. In catalytic processes, oxygen at or near the surface of e.g. CeO2 nanoparticles is believed to constitute (part of) the catalytically active sites,5 yet the identity and role of specific surface oxide environments in this and other nanoparticle systems remains uncertain.
Nuclear magnetic resonance (NMR) spectroscopy can reveal a wealth of chemical and structural information on the atomic scale, and previous work has shown that 17O solid-state NMR (ssNMR) spectroscopy is a powerful tool in investigating the structure and activity of zeolites,6 metal oxide nanoparticles,7,8 and other functionally relevant oxides.9 However, the inherent difficulty of attaining sufficient signal to noise in NMR spectroscopy is exacerbated for experiments on 17O, the only NMR-active nucleus of oxygen, as its low natural abundance (0.037%) leads to lower intensity and its quadrupolar character (I = 5/2) can result in additional spectral broadening.
The challenges of acquiring ssNMR spectra are further confounded when studying surface environments, as they typically constitute a small fraction of the sample. Nonetheless, in recent work by Wang et al., 17O ssNMR spectra of nanoparticulate CeO2 have been recorded and assigned to specific surface environments via a combination of surface-selective enrichment (with H217O) and density functional theory (DFT) calculations.8 However, in this case the surface-selective enrichment is only possible due to the high reactivity of ceria.
A more general approach to overcome the sensitivity problems inherent to ssNMR is the use of dynamic nuclear polarisation (DNP),10 which has seen a significant resurgence in recent years. In a typical DNP experiment, the sample is impregnated with radicals in a frozen glassy solvent, and the spin polarisation of the unpaired electrons on the radicals is transferred to the NMR-active nuclei via application of high-frequency microwave radiation. As the equilibrium polarisation of the electron is much greater than that of nuclei, NMR signal enhancements exceeding a factor of 200 have been achieved.11
DNP can be applied in two ways: in direct DNP, the nucleus of interest is directly polarised by the radicals, whereas in indirect DNP, 1H nuclei are first polarised and cross polarisation (CP) is then used to transfer the 1H polarisation to the nucleus of interest. The latter approach typically leads to larger enhancement factors and permits shorter recycle delays, but requires 1H nuclei embedded in the sample. In particular, indirect DNP has been used to record the 13C NMR spectra of surface organic species covalently incorporated into silica frameworks,12 the 17O NMR spectra of surface hydroxyl groups in mesoporous silica nanoparticle samples13 and the 27Al NMR spectra of surface sites in γ-alumina nanoparticles.14 Direct DNP has been used to record the 27Al NMR spectra of surface sites in mesoporous alumina–silica15 and the 17O NMR spectra of MgO.16 However, to our knowledge, direct DNP has not thus far been used to perform surface-sensitive 17O ssNMR spectroscopy.
In this work, CeO2 nanoparticles are investigated in order to establish the feasibility of surface-selective direct DNP 17O NMR. The CeO2 nanoparticles (Sigma Aldrich) were first investigated using transmission electron microscopy (TEM) (Fig. 1a); this showed a predominantly octahedral morphology with an average particle size of 11 ± 5 nm. Identification of the (111) fringes with a spacing of 3.12 Å revealed that the particles were dominated by (111) facets, the structure of which is shown in Fig. 1b.
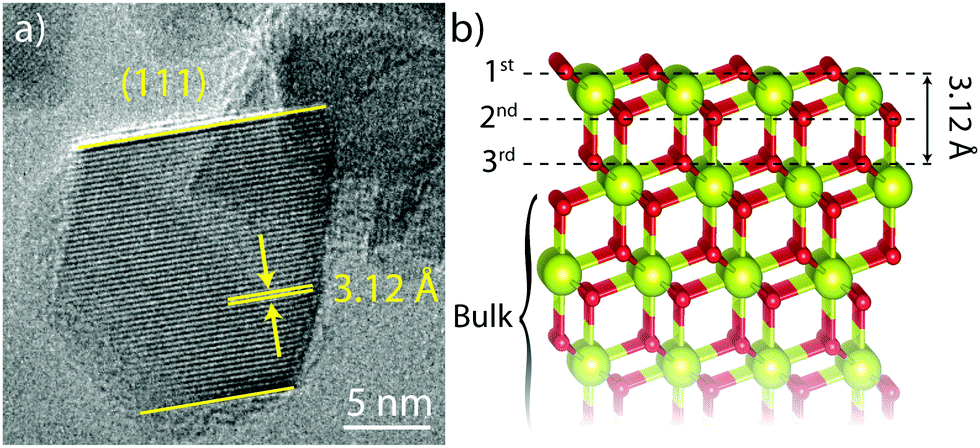 | ||
| Fig. 1 (a) HRTEM image of a CeO2 nanoparticle showing (111) fringes, (111) surfaces and an octahedral morphology (in projection), and (b) the structure of a (111) O-terminated CeO2 surface showing the first three oxygen layers and the (111) spacing. Cerium atoms are larger and yellow while oxygen atoms are smaller and red. | ||
The nanoparticles were then enriched with 17O2 (Cambridge Isotope Laboratories) at 350 °C for 24 hours and subsequently handled under an inert atmosphere (Ar or N2 gloveboxes) as the first surface layer readily exchanges with oxygen in the air, leading to loss of enrichment. TEM analysis of the enriched samples revealed minor coarsening with a subsequent average particle size of 15 ± 3 nm. To perform DNP experiments, the nanoparticles were wetted with the TEKPol biradical11 in 1,1,2,2-tetrachloroethane (TCE), and packed into a 3.2 mm sapphire rotor. This combination has been chosen rather than the alternative AMUPol/H2O,17 as the presence of un-enriched water can lead to removal of 17O from the first layer.8,1817O DNP NMR spectra were then recorded at 14.1 T under low temperature (∼95 K) magic angle spinning (MAS) using a pre-saturated Hahn-echo experiment, with a single rotor echo delay (100 μs). Fig. 2a shows the 17O ssNMR spectra recorded with and without microwave irradiation (“ON” and “OFF” respectively), with 8 scans and a recycle delay of 60 s. Without microwave irradiation, only the sharp signal due to the single bulk oxygen environment could be observed, whereas under microwave irradiation three new features were distinguished; these are ascribed to (sub-)surface sites selectively enhanced by TEKPol radicals in the vicinity of the surface.
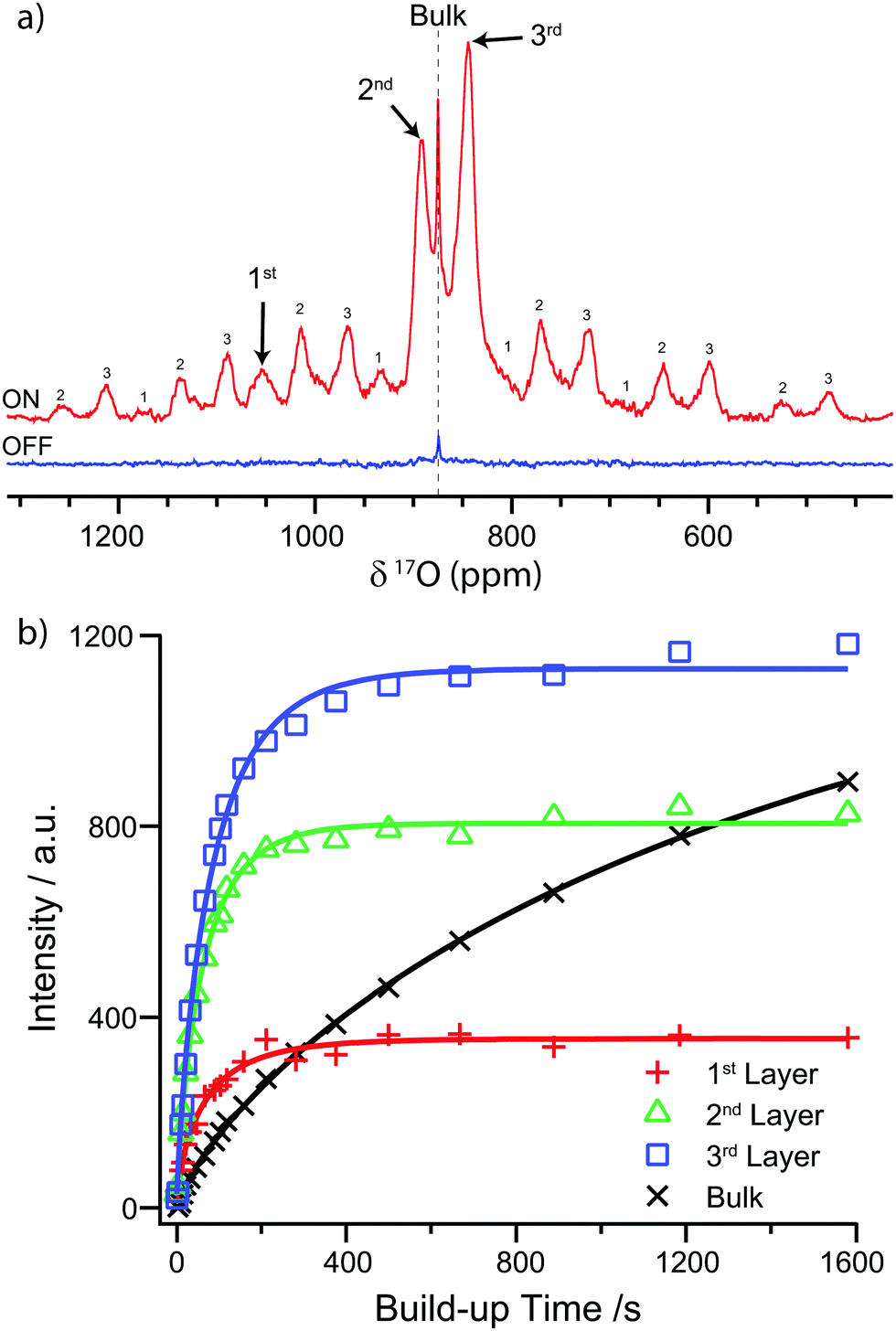 | ||
| Fig. 2 (a) 17O NMR (14.1 T) spectra of 17O enriched CeO2 nanoparticles mixed with the TEKPol radical in TCE, with and without microwave irradiation, using a presaturated Hahn echo experiment. The spectra were recorded at 95 K. The OFF spectrum was recorded at 12.5 kHz MAS, whereas the ON spectrum was recorded at 10 kHz in order to separate the spinning sidebands from the signal arising from the first layer. Spinning sidebands are labelled according to the layer of the signal from which they arise. (b) The 17O saturation recovery build-up curves for the different environments in CeO2 nanoparticles and the fitted stretched exponentials. The intensity is determined by deconvoluting the isotropic peaks (see Fig. S8 and discussion in ESI†). | ||
By comparison with the DFT calculations of Wang et al.,8 these features at 1055, 893, and 843 ppm are identified as oxygen sites within the first, second and third (sub-)surface layers, respectively (labelled in Fig. 1b). There is some discrepancy between the chemical shifts observed in this work and those reported by Wang et al., which is ascribed to minor differences between the CeO2 samples (see Table S1 and discussion in ESI†). The (sub-)surface sites also have a larger anisotropy than the bulk as evidenced by the greater intensity of their spinning sideband manifolds, consistent with the reduction of symmetry at the surface. The DFT calculations suggest that the sideband intensity predominately arises from satellite transitions which are broadened by the larger quadrupolar coupling constants (CQ) of the (sub-)surface sites (100–135 kHz for the first three layers cf. zero for the tetrahedral bulk sites); however, this alone does not fully account for the sideband intensity. Another contributing factor is the electron–nuclear dipolar coupling with the radicals; however, simulation of the sideband manifold suggests that bulk magnetic susceptibility effects due to the paramagnetic matrix19 must dominate (see Fig. S3 and discussion in ESI†). The origin of the broadening of the (sub-)surface signals observed with DNP can be identified by comparing the DNP spectrum to the conventional room temperature ssNMR spectra, with and without the addition of radicals (Fig. S5 and S1 respectively, ESI†), which shows that the broadness is caused by freezing out of motional averaging at ∼95 K, most likely of the radicals.
The observed surface selectivity in direct DNP occurs because the radicals are external to the particles and the rate of polarisation transfer from the radical to a nucleus falls off rapidly (as 1/r6).20 Nuclei at the surface can therefore be hyper-polarised by the radicals, but for sites within the deeper sub-surface layers, the excess nuclear spin polarisation must travel via spin diffusion, which is thought to be slow for 17O (in part due to the low natural abundance and gyromagnetic ratio),21 leading to a longer build-up time.
To test this hypothesis, the DNP build-up time constant (TDNP) was determined for each feature using a saturation recovery experiment (see ESI† for further details). The nuclear magnetisation was first nullified with a saturation pulse train and then allowed to build up via DNP for a variable time before measuring the resulting magnetisation by recording the 17O NMR spectrum. The build-up time was found by fitting the signal intensity to a stretched exponential function of the form

| Bulk | 1st Layer | 2nd Layer | 3rd Layer | |
|---|---|---|---|---|
| Shift/ ppm | 875 | 1055 | 893 | 843 |
| T DNP /s | >1600 | 67 ± 6 | 62 ± 2 | 85 ± 3 |
A second experiment has been performed on a sample of the same nanoparticles, but enriched with a higher pressure of 17O2 and stored under ambient conditions (Fig. S4, ESI†). For this sample, the second and third layer sites are again observed but the signal arising from the first layer is not observed due to exchange with 16O2 in air; this can be shown by re-recording the conventional ssNMR spectrum after progressive exposure to air and observing the concomitant reduction in signal for the first layer (Fig. S7, ESI†). The bulk signal is more intense than previously, which is ascribed to increased incorporation of 17O due to the larger 17O2 pressure during enrichment; the build-up time constant for the bulk signal is also smaller than the previous sample (TDNP = 586 s, Fig. S6, ESI†), which is ascribed to faster spin diffusion into the bulk due to the greater enrichment, because spin diffusion is strongly dependent on the concentration of the spin-active nucleus. The higher enrichment level also allows the (sub-)surface sites to be observed without DNP in a long (12 h) experiment (Fig. S5, ESI†), and hence DNP enhancements (εON/OFF) for the second and third layers can be measured as 56 and 29, respectively. The larger enhancement of the second layer is presumably due to less efficient hyperpolarisation of the more distant third layer (the quenching due to radicals is present with or without microwave irradiation, so does not affect the enhancement factor). The bulk site exhibits only a very minor enhancement as it is dominated by atoms far from the surface which are not hyperpolarised. We note, however, that as a recycle delay of 60 s is insufficient to obtain the maximum signal either with or without microwave irradiation, the observed enhancement factors for all sites will be dependent on the recycle delay. As has been previously addressed by Lee et al., εON/OFF should be seen as a guide to the DNP enhancement rather than a fundamental parameter.23 These results show that the DNP NMR spectra of CeO2 nanoparticles are sensitive to details of sample preparation, which can in turn give insight into the mechanisms of DNP.
To compare with these direct DNP experiments, indirect DNP (1H→17O) NMR spectra were also recorded on the first nanoparticulate CeO2 sample (Fig. 3). These revealed 17O signals centred at 225 ppm and −20 ppm, which are ascribed to Ce–OH terminations and H2O molecules adsorbed to the surface, respectively, again in agreement with Wang et al.8 These assignments are supported by the short CP contact time of 200 μs required to attain the maximum CP intensity, which is indicative of direct O–H bonding; the signal is attenuated with longer contact times. These signals could not be observed with direct DNP, even when the carrier frequency was varied and the field was swept to optimise the enhancements (Fig. S10, ESI†), but require the greater enhancement factors achievable with indirect DNP. The indirect DNP experiments do not, however, exhibit evidence for the (sub-)surface sites identified via direct DNP NMR, even under conditions of longer contact times and variable rf carrier frequencies (efficient CP is only observed to signals close to the carrier frequency). The lack of (sub-)surface oxygen features is attributed to the 1/r6 dependence of CP on distance and the difficulty of spin-locking the quadrupolar 17O nucleus, so that only oxygen atoms directly bonded to hydrogen can be readily seen.24 Furthermore, there are few hydroxyl terminations and adsorbed water molecules on surfaces of CeO2 samples oxidised at >300 °C,25 and the hydrophobic TCE solvent does not adsorb strongly, so insufficient protons exist in the vicinity of the surface to permit efficient CP. Therefore, direct DNP is needed in this case to observe surface and sub-surface oxygen sites in nanoparticulate CeO2.
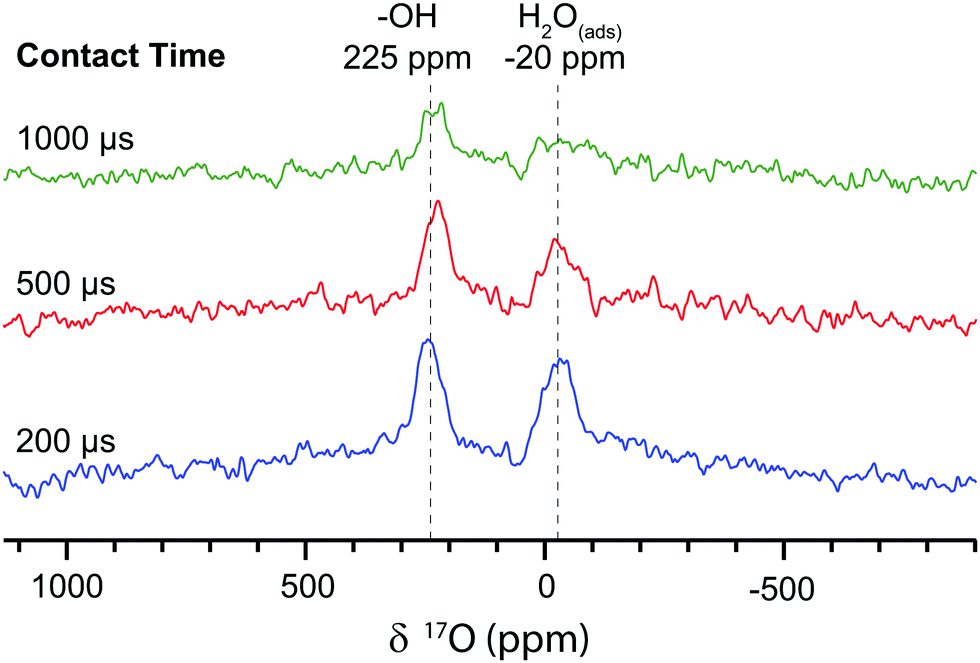 | ||
| Fig. 3 The indirect DNP 17O NMR (14.1 T) spectra of 17O enriched CeO2 nanoparticles impregnated with TEKPol in TCE, recorded at 12.5 kHz MAS with a recycle delay of 4.3 s, 320 scans and variable contact times for the 1H→17O cross polarisation. The 17O magnetisation was pre-saturated to avoid the direct DNP signal. | ||
Finally, to determine whether (sub-)surface signals could be used to distinguish between differing morphologies, CeO2 nanorods were also investigated by direct 17O DNP NMR (ESI,† Section 8); surface selectivity was again observed with a reduction in the intensity of the second sub-surface layer, which may indicate preferential segregation of oxygen vacancies.
In conclusion, surface-selective direct 17O DNP NMR spectroscopy has been demonstrated for the first time, using a system of CeO2 nanoparticles, for which the first three layers can be distinguished with high selectivity. This selectivity is ascribed to the slow spin diffusion of 17O polarisation into the bulk, so that only the (sub-)surface sites are efficiently hyper-polarised by radicals in the vicinity of the surface. This is corroborated by the build-up curves for the different signals and by comparison between samples with different degrees of enrichment. It is shown that although indirect DNP can be used to identify –OH terminations and adsorbed water on the CeO2 surface, it is not possible to observe the aforementioned (sub-)surface sites via this approach due to the scarcity of protons near the surface and the difficulty of long-distance 1H→17O cross polarisation; (sub-)surface sites can only be detected with direct DNP. The observed (sub-)surface signals can be used to distinguish between morphologies and we note that this approach may be extended to other systems where protons are not available to allow indirect DNP experiments, or where the surfaces are sensitive to water exposure.
We are grateful for financial support by the Oppenheimer Foundation (M. A. H.), the Cambridge Commonwealth Trusts (D. M. H.), the National Natural Science Foundation of China (NSFC) (21573103 and 21661130149) and the Royal Society Newton Fund (L. P.). The DNP experiments were performed at the DNP MAS NMR Facility at the University of Nottingham, with thanks to the EPSRC for funding of pilot studies (EP/L022524/1). We would also like to thank Dr D. A. Jefferson (Cambridge) for acquiring the TEM images and for useful discussions, Craig Stoppiello and Prof. A. N. Khlobystov (Nottingham) for use of their glovebox, Dr Xin-Ping Wu and Dr Xue-Qing Gong (ECUST, Shanghai) for the DFT calculations on CeO2 and Dr Yuxian Gao and Prof. Weixin Huang (University of Science and Technology of China) for synthesising the CeO2 nanorods. The experimental data for all NMR experiments have been made available at DOI: 10.17863/CAM.7267.
References
- M. Fernández-García and J. A. Rodriguez, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, 2011 Search PubMed.
- Y. Ren, Z. Liu, F. Pourpoint, A. R. Armstrong, C. P. Grey and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 2164–2167 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- L. Peng, Y. Liu, N. Kim, J. E. Readman and C. P. Grey, Nat. Mater., 2005, 4, 216–219 CrossRef CAS PubMed.
- E. Scolan, C. Magnenet, D. Massiot and C. Sanchez, J. Mater. Chem., 1999, 9, 2467–2474 RSC.
- M. Wang, X.-P. Wu, S. Zheng, L. Zhao, L. Li, L. Shen, Y. Gao, N. Xue, X. Guo, W. Huang, Z. Gan, F. Blanc, Z. Yu, X. Ke, W. Ding, X.-Q. Gong, C. P. Grey and L. Peng, Sci. Adv., 2015, 1, e1400133 Search PubMed.
- D. M. Halat, R. Dervişoğlu, G. Kim, M. T. Dunstan, F. Blanc, D. S. Middlemiss and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 11958–11969 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Coperet, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed.
- F. A. Perras, U. Chaudhary, I. I. Slowing and M. Pruski, J. Phys. Chem. C, 2016, 120, 11535–11544 CAS.
- V. Vitzthum, P. Miéville, D. Carnevale, M. A. Caporini, D. Gajan, C. Copéret, M. Lelli, A. Zagdoun, A. J. Rossini, A. Lesage, L. Emsley and G. Bodenhausen, Chem. Commun., 2012, 48, 1988 RSC.
- A. Lund, M.-F. Hsieh, T.-A. Siaw and S.-I. Han, Phys. Chem. Chem. Phys., 2015, 17, 25449–25454 RSC.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS PubMed.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef PubMed.
- Y. Champouret, Y. Coppel and M. L. Kahn, J. Am. Chem. Soc., 2016, 138, 16322–16328 CrossRef CAS PubMed.
- A. S. L. Thankamony, O. Lafon, X. Lu, F. Aussenac, M. Rosay, J. Trébosc, H. Vezin and J. P. Amoureux, Appl. Magn. Reson., 2012, 43, 237–250 CrossRef.
- K. R. Thurber and R. Tycko, J. Chem. Phys., 2012, 137, 84508 CrossRef PubMed.
- F. A. Perras, T. Kobayashi and M. Pruski, J. Am. Chem. Soc., 2015, 137, 8336–8339 CrossRef CAS PubMed.
- L. Holmes, L. Peng, I. Heinmaa, L. A. O;Dell, M. E. Smith, R.-N. Vannier and C. P. Grey, Chem. Mater., 2008, 20, 3638–3648 CrossRef CAS.
- D. Lee, S. Hediger and G. De Paëpe, Solid State Nucl. Magn. Reson., 2015, 66, 6–20 CrossRef PubMed.
- J. P. Amoureux and M. Pruski, Mol. Phys., 2002, 100, 1595–1613 CrossRef CAS.
- A. Laachir, V. Perrichon, A. Badri, J. Lamotte, E. Catherine, J. C. Lavalley, J. El Fallah, L. Hilaire, F. Le Normand, E. Quéméré, G. N. Sauvion and O. Touret, J. Chem. Soc., Faraday Trans., 1991, 87, 1601–1609 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, conventional ssNMR spectra, fitting of the sidebands, the effect of air exposure, DNP build-up curve analysis, comparison with CeO2 nanorods and the effect of a field sweep on the DNP enhancement. See DOI: 10.1039/c6cc10145c |
| This journal is © The Royal Society of Chemistry 2017 |