
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling and tuning the dynamic nature of supramolecular polymers in aqueous solutions†
Simone I. S.
Hendrikse
 a,
Sjors P. W.
Wijnands
a,
René P. M.
Lafleur
a,
Maarten J.
Pouderoijen
b,
Henk M.
Janssen
b,
Patricia Y. W.
Dankers
a and
E. W.
Meijer
*a
a,
Sjors P. W.
Wijnands
a,
René P. M.
Lafleur
a,
Maarten J.
Pouderoijen
b,
Henk M.
Janssen
b,
Patricia Y. W.
Dankers
a and
E. W.
Meijer
*a
aInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl; Tel: +31 040 2473101
bSyMO-Chem B.V., Het Kraneveld 14, 5612 AZ Eindhoven, The Netherlands
First published on 25th January 2017
Abstract
Structural and kinetic exchange properties of supramolecular polymers composed of mono- and bivalent ureidopyrimidinone-based monomers are investigated in aqueous solutions. It is shown that exchange dynamics can be controlled by mixing different types of monomers. This tunability widens the scope in their design as biomaterials.
Water-soluble supramolecular polymers constitute an attractive class of polymers, since the non-covalent interactions between the molecular components can result in tuneable hydrogels with a highly dynamic structure.1–3 When applied as a biomaterial, the dynamic, adaptable, and reversible interactions between the hydrogel and embedded (stem) cells are proposed to closely mimic systems and processes found in nature. The artificial biomaterial developed shall not only contain specific mechanical properties, but also has to present proteins and peptides to cells in a reversible, adaptive and spatiotemporal manner in order to regulate cell response.4,5 Therefore, control over the structural and dynamic properties of hydrogels is important for the design and development of supramolecular biomaterials, which can only be achieved when there is profound understanding at the molecular level.
Many techniques are currently used to study such supramolecular systems in aqueous solutions.6 Förster resonance energy transfer (FRET) experiments7,8 have been used to study exchange dynamics in a broad spectrum of systems from liposomes9 to polymers. In addition, fluorescence microscopy, and more recently super resolution microscopy techniques such as stochastic optical reconstruction microscopy (STORM) yield valuable structural and temporal information of systems ranging from giant unilamellar vesicles to 1D-supramolecular aggregates such as benzene-1,3,5-tricarboxamide based polymers and peptide amphiphiles.10–12 Furthermore, stimulated emission depletion (STED) microscopy has been successfully applied in studying the formation and characteristics of multicomponent self-sorted supramolecular hydrogels in real-time.13
Using molecular design parameters to tune the macroscopic properties of hydrogels has been nicely shown by Scherman and Stupp. Scherman has shown that different guests for cucurbit[8]uril mediated crosslinking resulted in distinct crosslink dynamics, influencing the mechanical properties at the hydrogel level.14 Stupp has revealed that a disordered fibre packing can be obtained by substituting valine for alanine in peptide amphiphiles, which subsequently decreases the physical properties of the hydrogel.15
Here we have investigated the structural and kinetic properties at the nanometer scale of supramolecular ureidopyrimidinone (UPy) based polymers in water. The UPy moiety is a self-complementary unit containing a quadruple hydrogen bonding array. In chloroform the high binding constant of 6 × 107 M−1 results in a bonding life-time of roughly one second.16 In order to obtain similar values in water, the UPy unit requires hydrophobic shielding from water-soluble oligo- and poly(ethylene glycol) groups.17 Additionally, urea groups are introduced in the hydrophobic alkyl spacers to enable lateral stacking into long 1D-polymers due to a combination of hydrophobic effects, π–π stacking and hydrogen bonding. Recently, we have shown that both mono- and bivalent UPy monomers assemble into supramolecular polymers that give rise to hydrogels at concentrations of only a few weight percentage (wt%).17 By mixing the mono- and bivalent UPy monomers, hydrogels are obtained of which their stiffness can be controlled by the composition of the mixture.18
The structural and temporal properties of the assembly of monovalent monomers mUPy 1 and bivalent monomers bUPy 2 (Scheme 1) in water has been elucidated using cryogenic transmission electron microscopy (cryo-TEM), STORM and FRET experiments. For the latter two techniques, 2 mol% of dye-labelled probes (monovalent mDye 3a and mDye 3b, and bivalent bDye 4a and bDye 4b; Scheme 1) were embedded in the aggregates formed by monomers 1 and 2.
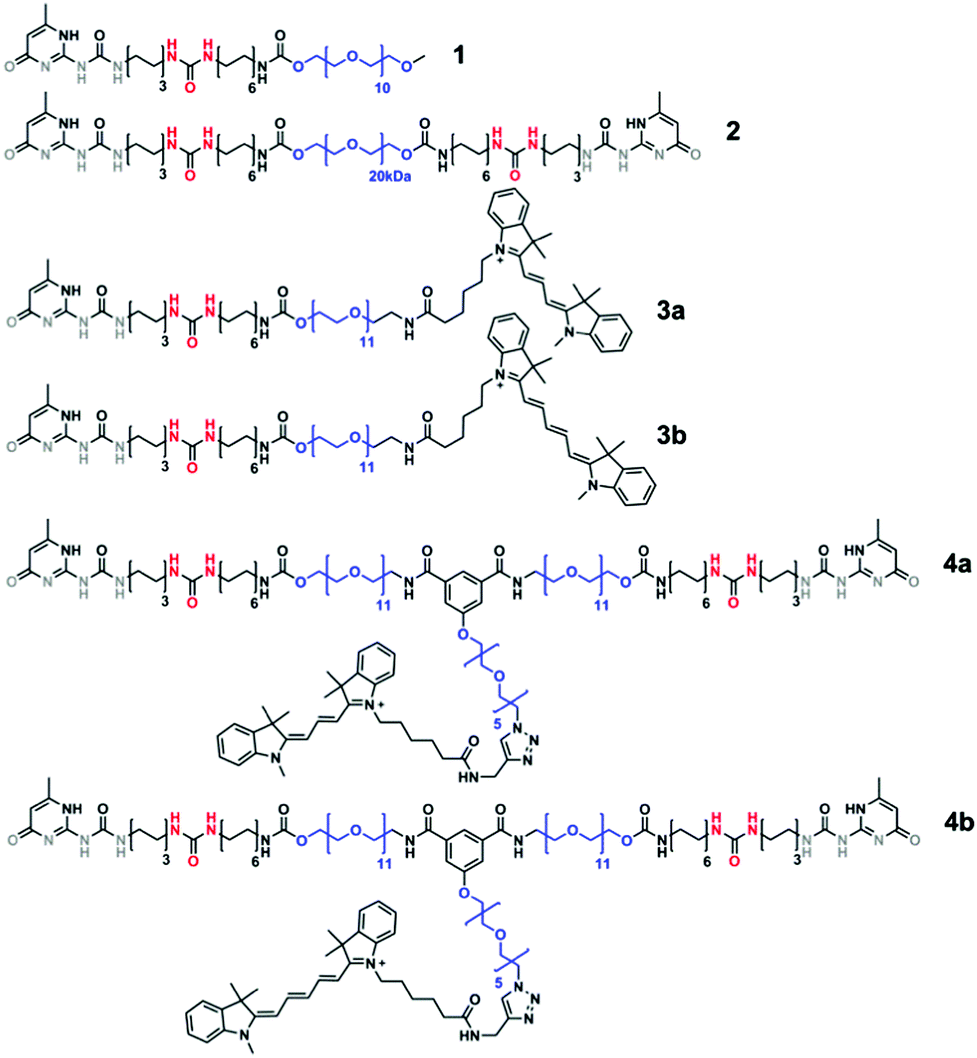 | ||
| Scheme 1 Molecular structures of monovalent mUPy 1, bivalent bUPy 2, dye-labelled monovalent UPy mDye 3a and mDye 3b, and dye-labelled bivalent UPy bDye 4a and bDye 4b. The scaffolding materials 1 and 2 are studied in combination with 2 mol% of dyes 3 and 4 that serve as molecular probes. The UPy based molecules dimerize via quadruple hydrogen bonding arrays (grey) and laterally stack via the urea moieties (red). Poly(ethylene glycol) spacers (blue) provide water solubility. | ||
Solutions of 1 and 2 were stirred in water at 72 °C for 1.5 hours, and were then slowly cooled to room temperature. Subsequently, the structural characteristics of the scaffolds formed by 1 and 2 were compared using cryo-TEM. Micrometre long fibrillar structures with diameters of approximately 5 and 14 nm were observed for 1, while shorter fibrillar structures in the range of hundreds of nanometres with an apparent diameter of approximately 7 nm were found for 2 (Fig. 1 and ESI,† Fig. S1). The 14 nm diameter of 1 corresponds to bundled dimers, whereas the 5 nm of 1 and 7 nm of 2 are proposed to correspond to single dimers. Please note that the water shell surrounding the PEG part of the molecules does not scatter sufficient electrons, and in the case of 2, the longer PEG surrounding the fibre backbone sequesters more water, blurring the contrast of the inner hydrophobic part. The incorporation of the dye-labelled monomers 3a, 3b, 4a, and 4b within these fibres was investigated using STORM. The measurements were performed at 12.5 μM sample concentrations, using 1 or 2 in combination with 2 mol% of the dye-labelled probes 3a, 3b, 4a, or 4b. Micrometre long fibre scaffolds were observed for 1 (with mDye 3b, see Fig. 2, left), corroborating the cryo-TEM results for 1. In contrast, smaller, less-defined 1D-aggregates were observed for 2 (with mDye 3b, see Fig. 2 right) with a maximal length of a few hundreds of nanometres. Despite the >20 fold poorer spatial resolution of the technique,19 the ∼40 times lower concentration, and/or effects of sample preparation and adsorption on the glass surface, these structures formed by 2 appeared relatively similar as compared to the features observed using cryo-TEM. Nevertheless, the results of the two techniques showed a clear structural difference between the aggregates formed by the two UPy derivatives, where the long fibres of 1 and the shorter and less-defined fibres of 2 indicate a differentiation in the packing of the monomers.
 | ||
| Fig. 1 Cryo-TEM images of scaffolds formed by monovalent mUPy 1 (left) and bivalent bUPy 2 (right). Fibrillar structures were observed in both cases with a diameter of approximately 5 and 14 nm for mUPy 1 (c = 0.5 mg ml−1; 482 μM) and approximately 7 nm for bUPy 2 (c = 10 mg ml−1; 474 μM). The scale bars represent 100 nm (left) and 200 nm (right). | ||
 | ||
| Fig. 2 STORM images of scaffolds formed by monovalent 1 (left) and bivalent 2 (right) with 2 mol% of mDye 3b incorporated (cTotal = 12.5 μM). Monovalent dye-labelled UPy 3b was incorporated into both scaffolds, showing micrometre long fibres for mUPy 1 and fibres of a few hundreds of nanometres for bUPy 2. The scale bars represent 1 μm. | ||
Next, the excellent FRET pair, donor Cy3 and acceptor Cy5,20 were selected for studying the exchange of monomeric units in and between 1D-fibre scaffolds. Separate solutions containing Cy3 (mDye 3a or bDye 4a) and Cy5 (mDye 3b or bDye 4b) were prepared by incorporating 2 mol% of these dye-probes 3a, 3b, 4a or 4b into pre-assembled scaffold solutions containing 1 or 2. Specifically, a 24.5 μM scaffold and 0.5 μM dye probe concentrations were used (sample preparation described in the ESI†). Subsequently, the corresponding Cy3 and Cy5 solutions (i.e.3a and 3b, or 4a and 4b) were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Upon excitation of the donor, the fluorescence of both dyes was measured (Fig. 3). It is known that a close proximity (2–10 nm) of both dyes results in efficient energy transfer.8 Because of the non-covalent interactions between the monomers, the dye-labelled monomers can migrate within and between fibres. As a result, this technique investigates the exchange dynamics of the dye-labelled monomers within and between fibres. Two distinct exchange rates for the two scaffolds 1 and 2 were observed (Fig. 3 and ESI,† Fig. S2): immediately after mixing, a very efficient FRET was observed for mDyes 3a and 3b incorporated in 2 (Fig. 3, upper left), whereas only a low FRET was observed for mDyes 3a and 3b incorporated in 1 (Fig. 3, lower left). When increasing the temperature, the exchange of 1 was accelerated, even though the energy transfer did not reach a maximum, as was observed with 2 (ESI,† Fig. S3). Comparing the exchange rates of monovalent mDyes 3 and bivalent bDyes 4 within the same scaffold, a slower exchange profile for 4 was observed (Fig. 3, compare left and right). This is hypothesized to be due to multivalency effects. In addition, the exchange dynamics of 4 embedded in 2 were further reduced when the FRET experiments were performed with aged samples (ESI,† Fig. S4).
:1 ratio. Upon excitation of the donor, the fluorescence of both dyes was measured (Fig. 3). It is known that a close proximity (2–10 nm) of both dyes results in efficient energy transfer.8 Because of the non-covalent interactions between the monomers, the dye-labelled monomers can migrate within and between fibres. As a result, this technique investigates the exchange dynamics of the dye-labelled monomers within and between fibres. Two distinct exchange rates for the two scaffolds 1 and 2 were observed (Fig. 3 and ESI,† Fig. S2): immediately after mixing, a very efficient FRET was observed for mDyes 3a and 3b incorporated in 2 (Fig. 3, upper left), whereas only a low FRET was observed for mDyes 3a and 3b incorporated in 1 (Fig. 3, lower left). When increasing the temperature, the exchange of 1 was accelerated, even though the energy transfer did not reach a maximum, as was observed with 2 (ESI,† Fig. S3). Comparing the exchange rates of monovalent mDyes 3 and bivalent bDyes 4 within the same scaffold, a slower exchange profile for 4 was observed (Fig. 3, compare left and right). This is hypothesized to be due to multivalency effects. In addition, the exchange dynamics of 4 embedded in 2 were further reduced when the FRET experiments were performed with aged samples (ESI,† Fig. S4).
 | ||
| Fig. 3 Emission scans recorded after mixing solutions of mDye 3a and 3b or bDye 4a and 4b incorporated in scaffolds formed by mUPy 1 or bUPy 2 at different time points (cScaffold = 24.5 μM, cDye = 0.5 μM, excitation at 540 nm). Immediate exchange occurred in samples containing bUPy 2 whereas a very low FRET was observed in samples containing mUPy 1. | ||
Finally, monovalent scaffold 1 and bivalent scaffold 2 were co-assembled in different ratios in order to investigate the effect of fibre composition on the exchange dynamics (Fig. 4 and ESI,† Fig. S5). Upon increasing the content of 2 with respect to 1, both the total exchange and the exchange rate were increased. The same trend was observed for both dyes, mDyes 3 and bDyes 4, however, a slightly lower FRET was observed with 4 when comparing the same fibre composition. Analysing the FRET experiments all-together, the dye-labelled monomers incorporated in 2 were more dynamic than in 1. Both the scaffold composition and the bivalency of the dye-labelled monomer influence the exchange dynamics. However, the scaffold composition dominates the kinetic properties, whereas the bivalency effect of the dye-labelled monomer is small. These observations indicate that the packing of the monomers is extremely important.
 | ||
| Fig. 4 Co-assembly of monovalent scaffold 1 with bivalent scaffold 2, incorporated with 2 mol% of bivalent dyes 4 (cTotal = 25 μM). Left: Normalized Cy3 intensity; right: normalized intensity of the Cy5 peak upon Cy3 excitation, indicating FRET. Upon increasing the bivalent scaffold 2 content, the exchange dynamics were increased due to a disordering of the monomer packing. | ||
Since FRET is an ensemble technique and therefore only provides averaged data, the exchange dynamics of the dye-labelled monomers between individual fibres was visualized employing STORM.11 For this, the samples, as prepared for the FRET experiments, were diluted to 12.5 μM with PBS and mixed in a 1:1 ratio with its corresponding variant (i.e. Cy3 solution a, 3a or 4a, mixed with Cy5 solution b, 3b or 4b, respectively). The fibres were adsorbed on coverslips at different time points. Afterwards, dual channel imaging was performed where both Cy3 (green) and Cy5 (red) were excited simultaneously. Monovalent dyes 3a and 3b embedded in monovalent scaffold 1 after 1 minute of mixing (t = 1 min) revealed fibres which were predominantly green or predominantly red, indicating that negligible exchange had taken place (Fig. 5, upper row). However, after 20 hours, a small amount of random exchange was observed as a few red labels were observed in predominantly green fibres and vice versa. In contrast, exchange of the monovalent dyes 3a and 3b in 1D-fibers of bivalent scaffold 2 occurred immediately after mixing, indicated by both colours (i.e. green and red) being within the same fibre. Moreover, imaging of the mixed fibre (i.e. scaffolds containing 50% 1 and 50% 2) showed fibres of intermediate lengths, demonstrating successful mixing and ruling out any self-sorting of 1 and 2 (ESI,† Fig. S6).
 | ||
| Fig. 5 STORM images of scaffolds formed by monovalent 1 (top and middle row) and bivalent 2 (bottom row) with 2 mol% of incorporated mDye 3a + 3b (cTotal = 12.5 μM). Left: Merged channel, middle: Cy5 excited channel, and right: Cy3 excited channel. Directly after mixing mDye 3a and 3b incorporated in monovalent scaffold 1, negligible exchange was observed (top, t = 1 min), and after 20 hours a small degree of exchange was observed (middle, t = 20 hours). When the dyes were incorporated in bivalent scaffold 2, instant exchange was observed (bottom, t = 1 min). The scale bars represent 2 μm. | ||
By using a combination of powerful techniques, the structural and temporal properties of UPy based supramolecular polymers were investigated on the molecular level. This revealed a clear difference between fibres formed by monovalent scaffold 1 and bivalent scaffold 2. The long, well-defined fibres formed by 1 exhibited slow dynamics and even seemed to be near static. This is in sharp contrast with the structures formed by 2, which showed only short fibres and proved to have a fast exchange profile. These results clearly show the impact of the molecular design on the microscopic structures that the molecules are forming. The large ethylene glycol chain in 2 (∼454 vs. 10 repeating units in 1) destabilizes the self-assembly of the monomers due to steric and entropic effects, preventing an ordered packing and hence hampering the formation of well-defined fibres. The destabilization of supramolecular polymers by steric effects was also observed by Besenius et al.21 Furthermore, mixing of the UPy scaffolds formed by 1 and 2 resulted in destabilization of the ordered fibres upon an increasing content of 2, which subsequently increased the exchange rate of the monomers. It is shown that the dynamics can be tuned by adjusting the fibre composition, predominantly dominated by a difference in packing. Mixing scaffold 1 in similar fibres as 2 was previously shown to improve the stiffness at the macroscopic level significantly. This was proposed to be due to a difference in exchange rates at the molecular level.18 Here we confirm that the exchange rates of the dye-labelled monomers are dominated by the scaffold type, and to a lesser extent by the multivalency of the dye-labelled monomer. Moreover, the hydrophobic nature of the dyes might result in a preference to reside in the hydrophobic pocket of the fibre. Although no clear differences in fibre formation were observed with cryo-TEM and UV-vis (ESI,† Fig. S7), changes in the exchange kinetics are likely to occur.22
Despite it remaining a challenge to unravel the exact molecular structure and kinetics of supramolecular systems, cryo-TEM, FRET and STORM proved to be a powerful combination of techniques to study supramolecular fibres in great detail. The results indicate that both the structure and dynamics of these fibres heavily depend on the design of the UPy monomers, and we showed that it is possible to control their properties by mixing the different monomer variants. These are important findings for the design of functional monomers such as bioactive peptides and proteins introduced into supramolecular fibres, since they might change the packing within the fibres, and as a consequence, the macroscopic properties. Because there is a delicate balance between dynamic spatiotemporal presentation and the ability of the cell to locally remodel the surrounding microscopic environment, we anticipate that the studies described here are useful for the design of biomaterials.
This work was supported by the European Research Council (Grant Agreement 246829), the Royal Netherlands Academy of Science, the Netherlands Organization of Sciences (NWO-Newpol), and the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035).
Notes and references
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- H. J. Anderson, J. K. Sahoo, R. V. Ulijn and M. J. Dalby, Front. Bioeng. Biotechnol., 2016, 4, 38 Search PubMed.
- M. Akhmanova, E. Osidak, S. Domogatsky, S. Rodin and A. Domogatskaya, Stem Cells International, 2015, 2015, 35 CrossRef PubMed.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- A. K. Kenworthy, Methods, 2001, 24, 289–296 CrossRef CAS PubMed.
- N. D. Huebsch and D. J. Mooney, Biomaterials, 2007, 28, 2424–2437 CrossRef CAS PubMed.
- S. W. A. Reulen and M. Merkx, Bioconjugate Chem., 2010, 21, 860–866 CrossRef CAS PubMed.
- N. Momin, S. Lee, A. K. Gadok, D. J. Busch, G. D. Bachand, C. C. Hayden, J. C. Stachowiak and D. Y. Sasaki, Soft Matter, 2015, 11, 3241–3250 RSC.
- L. Albertazzi, D. van der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. van der Hofstad and E. W. Meijer, Science, 2014, 344, 491–495 CrossRef CAS PubMed.
- R. M. P. da Silva, D. van der Zwaag, L. Albertazzi, S. S. Lee, E. W. Meijer and S. I. Stupp, Nat. Commun., 2016, 7, 11561 CrossRef CAS PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS PubMed.
- E. A. Appel, R. A. Forster, A. Koutsioubas, C. Toprakcioglu and O. A. Scherman, Angew. Chem., Int. Ed., 2014, 53, 10038–10043 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- R. E. Kieltyka, A. C. H. Pape, L. Albertazzi, Y. Nakano, M. M. C. Bastings, I. K. Voets, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS PubMed.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- R. B. Mujumdar, L. A. Ernst, S. R. Mujumdar, C. J. Lewis and A. S. Waggoner, Bioconjugate Chem., 1993, 4, 105–111 CrossRef CAS PubMed.
- R. Appel, J. Fuchs, S. M. Tyrrell, P. A. Korevaar, M. C. A. Stuart, I. K. Voets, M. Schönhoff and P. Besenius, Chem. – Eur. J., 2015, 21, 19257–19264 CrossRef CAS PubMed.
- M. B. Baker, R. P. J. Gosens, L. Albertazzi, N. M. Matsumoto, A. R. A. Palmans and E. W. Meijer, ChemBioChem, 2016, 17, 207–213 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, methods, and supporting data of cryo-TEM, FRET, STORM and UV-vis experiments. See DOI: 10.1039/c6cc10046e |
| This journal is © The Royal Society of Chemistry 2017 |