
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular control over colloidal assembly
M.
Gerth
 a and
I. K.
Voets
*b
a and
I. K.
Voets
*b
aLaboratory of Physical Chemistry, and Institute of Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MD, Eindhoven, The Netherlands
bLaboratory of Physical Chemistry, Laboratory of Macromolecular and Organic Chemistry, and Institute of Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MD, Eindhoven, The Netherlands. E-mail: i.voets@tue.nl
First published on 20th March 2017
Abstract
Contemporary chemical and material engineering often takes inspiration from nature, targeting for example strong yet light materials and materials composed of highly ordered domains at multiple different lengthscales for fundamental science and applications in e.g. sensing, catalysis, coating technology, and delivery. The preparation of such hierarchically structured functional materials through guided bottom-up assembly of synthetic building blocks requires a high level of control over their synthesis, interactions and assembly pathways. In this perspective we showcase recent work demonstrating how molecular control can be exploited to direct colloidal assembly into responsive materials with mechanical, optical or electrical properties that can be adjusted post-synthesis with external cues.
 M. Gerth | Marieke Gerth graduated in Physical Chemistry from Wageningen University (The Netherlands) in 2015. She is currently a PhD candidate in the Laboratory of Physical Chemistry and the Institute for Complex Molecular Systems at Eindhoven University of Technology (The Netherlands) and works under the supervision of Prof. Dr Ir. R. Tuinier and Dr Ir. I. K. Voets. Her research focuses on the design, synthesis and characterisation of responsive supramolecular colloids. |
 I. K. Voets | Dr Ilja K. Voets studied Molecular Sciences at Wageningen University in Wageningen, the Netherlands. She performed her PhD research on complex coacervate core micelles assembled from oppositely charged copolymers at the same university, after which she moved to the Adolphe Merkle Institute in Fribourg, Switzerland for a postdoctoral stay. In 2011 she was appointed as Assistant Professor at the Department of Chemical Engineering and Chemistry and the Institute for Complex Molecular Systems of Eindhoven University of Technology (TU/e). Currently she is Associate Professor in the Laboratory of Macromolecular and Organic chemistry and the Laboratory of Physical Chemistry of the TU/e, where she leads an independent research group focusing on self-organised and bioinspired soft matter. |
1. Introduction
Nature's finest materials are hierarchical assemblies of a variety of building blocks organised in intricate architectures with length scales that span orders of magnitude from the atomic to granular level. The properties of these materials are as dependent on the chemical nature of their building blocks as on their spatiotemporal arrangement. Understanding and controlling the structural make-up and thereby properties of materials is thus one of the grand challenges in contemporary chemical and materials science. Co-organisation of multiple components through a combinatory interplay of covalent and supramolecular interactions both in- and out-of-equilibrium offers an elegant, low-energy route towards spatiotemporal control over hierarchically structured materials with emergent optical, mechanical, thermal, and electronic properties to name but a few.In this feature article we present and compare strategies to realise molecular control over colloidal assembly to create hierarchically structured metamaterials for fundamental studies and applications in sensing, catalysis, coating technology, and optoelectronics. Throughout, we will make a distinction between ordered supracolloidal structures, here termed ‘assemblies’, and disordered structures, here termed ‘aggregates’.
2. Colloidal stability and surface forces
We focus here on colloidal dispersions, which are fine mixtures of two immiscible phases, a solid discontinuous phase (i.e., colloidal particles) of which at least one dimension ranges from 1 nm to 10 μm in a liquid continuous phase. This nano- to mesoscopic size regime is viewed as the ‘colloidal domain’ bracketed on the lower limit by molecular length scales and on the upper limit by micro- and macroscopic dimensions of granular systems.Achieving predictable molecular control over colloidal assembly necessitates a detailed understanding and precise modulation of the surface forces that determine how particles interact with each other. Surface forces derive from intermolecular forces and are characterised by a range and depth (Fig. 1). For example, we quantify the range of electrostatic interactions in terms of the Debye length κ−1, which corresponds to the distance z from the particle surface where the interaction strength has decreased by 1/e. For short-ranged molecular interactions to be dominant, other surface forces operative between colloids must be tuned, typically weakened in strength and shortened in range.
 | ||
| Fig. 1 Colloidal surface forces. The strength of the repulsive electrostatic interactions (dashed grey line) and attractive van der Waals interactions (dashed black line) is strongly dependent on the surface-to-surface separation D. Together, these interactions often form an effective interaction potential U with a repulsive barrier and a secondary minimum (black line, square). The strength of the electrostatic repulsion is strongly dependent on the salt concentration so that the effective interaction can be strongly repulsive at low salt concentration (grey dash-dotted line, circle), or purely attractive at high salt concentration (grey line, triangle). | ||
Theoretically, ‘hard spheres’ represent the simplest class of colloidal particles. These non-deformable particles that cannot overlap exhibit an infinitely strong repulsive interaction at contact (when the surface-to-surface separation D = 0), or no interaction at all (at D > 0). Such colloids are said to interact solely through excluded-volume interactions. In silico, this is easy to achieve, but in practice, repulsive forces are not infinitely strong, while attractive interactions are difficult to suppress.
An omnipresent and often strong attraction is the van der Waals interaction with a typical range of a few nm, which is caused by a difference in polarizability between the colloid and the suspending medium.1 To diminish the van der Waals interaction experimentally, one can match the polarizability of the continuous and discontinuous phases by matching their refractive indices. In addition, colloids are often coated with a dense layer of small molecules or polymers to mask the remaining weakened attraction by steric hindrance. When this monolayer or polymer brush is thick enough, it simply makes the short separations corresponding to non-negligible van der Waals attraction inaccessible. Two widely studied hard-sphere-like colloidal model systems prepared in this manner are stearyl alcohol grafted silica colloids in cyclohexane2,3 and poly(methyl methacrylate) (PMMA) particles grafted with poly(12-hydroxystearic acid) in a mixture of tetralin, cis-decalin and carbon tetrachloride,4,5 or decalin and tetrachloroethylene.6
Alternatively, attractive van der Waals interactions can be counterbalanced in aqueous dispersions by repulsive screened electrostatic interactions. Colloids that carry a net charge due to e.g. ionisation of surface groups, ion adsorption or inclusion of charged initiators7–9 are surrounded by an ‘ion cloud’, which is a region of only a few Ångstroms thick with more counter-ions (ions with charge sign opposite to that of colloid) than co-ions, termed the ‘electrochemical double layer’, so that the colloid surrounded by its double layer is electroneutral. Close approach of two charged colloids thus requires ionic clouds to overlap, which generates an increased local osmotic pressure that pushes the colloids apart. Both the range and strength of this repulsive interaction can be tuned easily by variations in the salt concentration in the bulk, which impacts the electrolyte concentration gradient in the double layer. To maximise colloidal stability, aqueous colloidal dispersions are often electro-sterically stabilised by a neutral polymer brush with charged initiators, or by a charged polyelectrolyte brush.7,10 Orchestrating colloidal assembly with small molecules requires instead a sufficiently shallow interaction potential U(D) ∼ kT (be it net repulsive or attractive) at all separations D to ensure both access to and dominance of the short-ranged intermolecular forces.
3. Colloidal assembly
Since colloidal stability implies the absence of (un)controlled clustering, creaming and sedimentation, we must seek out conditions of borderline colloidal stability to induce assembly. This means that precise control over the delicate interplay of attractive and repulsive forces is of paramount importance.10,11 If interactions are too strong, bonds remain permanent and colloids cannot rearrange into a more thermodynamically favourable configuration. If interactions are too weak, assembly is absent or ensembles may fall apart. Ideally, one would by design eliminate all interactions except a (soft) repulsion operative only at contact and subsequently introduce a net attractive interaction tuneable in range and depth, so as to induce colloidal assembly in a reliable fashion.In 1954, Asakura and Oosawa showed that this is possible through so-called ‘depletion interactions’,12,13 which have since been studied and exploited for various purposes (see ref. 14 for a detailed account). Simply put, attractive depletion interactions arise in colloid/polymer (and colloid/colloid) mixtures as one species restricts the volume that can be occupied by the other, non-adsorbing species. Hence, the total excluded volume decreases if these inaccessible regions, the ‘depletion layers’ overlap, which causes an effective attraction due to a local imbalance in osmotic pressure. Interestingly, the strength and range of depletion forces can be tuned independently by variation of the concentration and molecular weight (hence size) of the polymer, as these determine the osmotic pressure and thickness of the depletion layer. Nowadays, there is widespread interest in depletion forces as a means to induce clustering into colloidal ensembles.14–19 Elegant examples of its effectiveness and robustness, particularly when combined with clever design of colloids with e.g. shape anisotropy or surface anisotropy, include the formation of ‘lock-and-key colloids’, ‘colloidal micelles’, and ‘colloidal polymers’.17–19
Electrostatically driven complexation between oppositely charged particles7,20 or complementary hydrogen bond donor- and acceptor-functionalised particles21 offers an attractive alternative route to induce colloidal assembly. Sparse yet percolating networks of clay platelets have been created from LAPONITE®, which is anisotropic in shape and non-uniform in charge distribution at low pH values, where the faces are negatively charged while the edges are positively charged.22,23 This leads to ‘edge-to-face’ assembly, which can be placed under molecular control through dissolution of a photoacid generator (PAG) into the aqueous dispersion, which lowers the dispersion pH upon UV irradiation.
Assembly into highly ordered (2D) structures via directional interactions generated by purely non-directional surface forces is one of the fascinating opportunities patchy particles offer.24–27 For example, a 2D Kagome lattice (Fig. 2) was built-up from spherical, electrostatically stabilised colloids coated with hydrophobic patches at their poles.27 Upon particle sedimentation and subsequent salt addition, the attractive interaction between the hydrophobic poles outweighed the screened electrostatic repulsion between the ‘bare’ equators, which induced assembly into a Kagome lattice. Clearly, control over patch number, patch size, specificity and interaction strength of the patchy (anisotropic) colloids is paramount to reliably construct assemblies with complex architectures.24–29
 | ||
| Fig. 2 Formation of a 2D colloidal Kagome lattice. (a) Particles with hydrophobic patches with an opening angle of 65° at the poles and charged equators, are allowed to sediment in deionised water. Addition of NaCl screens the electrostatic repulsion between the equators and allows self-assembly through hydrophobic interactions between the poles. (b) Fluorescence microscopy image of the resulting 2D Kagome lattice (main image) and its fast Fourier transform image (bottom right). Scale bar is 4 μm. (c) Enlarged view of the dashed white square in (b). The dashed red lines indicate two staggered triangles. (d) Schematic representation of particle orientations in the lattice. Adapted by permission from Macmillan Publishers Ltd: nature ref. 27, copyright 2011. | ||
4. Molecular control over colloidal assembly
The success of fine-tuning surface forces for colloidal assembly into supracolloidal architectures is demonstrated by the experimental realisation of colloidal micelles, plastic crystals, and 2D Kagome lattices to name but a few of the illustrative examples taken from recent literature.17,27,30,31 Establishing molecular control over colloidal association offers new and exciting opportunities such as spatiotemporal control over the structure and properties of colloidal materials (vide infra), but also poses additional constraints on the experimental design. Through (site-specific) functionalisation of the colloidal surface with self-assembling molecular moieties, the association at the colloidal lengthscale can be controlled by the small molecules tethered to the surface. The range of the attraction is now a few nm's at most – the range of the molecular interactions – instead of tuneable from a few nm's up to a few hundreds of nm's.9,18,32,33 Additionally, surface forces tend to be strong at such short separations, which constitutes a formidable challenge in adequately diminishing these non-specific interactions. Inevitably, effective strategies for molecular control therefore utilise a combinatorial approach to decrease the strength and/or alter the range of the ‘competing’ surface forces on the one hand and introduce externally addressable molecular control on the other hand.Strategies to diminish and/or modulate the range of surface forces have been much investigated. These include e.g. reduction of attractive van der Waals forces by matching refractive indices, salt addition to modulate the strength and range of repulsive screened Coulomb interactions, polymer addition to induce depletion attraction, and (non-)covalent attachment of molecular monolayers or polymer brushes to prohibit direct contact and establish a soft repulsion. Of these, particularly attractive are strategies that offer simultaneously a means to introduce molecular control. For example, colloids can be covered with a polymer brush comprising polymers end-functionalised with a photo-responsive, supramolecular, or metal-coordination-based recognition moiety.30,34,35
Once the surface forces are fine-tuned for molecular control, we can unlock the potential of vast libraries of small responsive molecules which are under continuous scrutiny and development to meet a plethora of requirements for various scientific and technological purposes. Optimal molecular guides for colloidal assembly fulfil all of the following criteria: solvophilicity, responsivity, external addressability, synthetic versatility, reactivity, detectability, stability, tuneability, reversibility, specificity and directionality. Versatility of synthesis, purification, and characterization is an obvious advantage as multi-step synthesis (and purification) routes are laborious and time-consuming, impeding the preparation of large amounts of material as well as regular optimisation of molecular structure and properties. Furthermore, the moieties must include a functionality for (verification of) surface-tethering and remain stable against degradation under the reaction and experimental conditions. All other requirements boost the potential for control over association into target structures. Defects can be repaired (locally) only if assembly is reversible and interactions (temporarily) weak. This is achievable through supramolecular bonds of tuneable strength which are responsive to, and preferably switchable by, external cues, like light, which can be spatiotemporally modulated. Reliable organisation in complex environments composed of mixtures of various functional colloidal species necessitates selective (like host–guest) interactions between specific compounds, which means aspecific attractive interactions due to e.g. solvophobicity must be avoided. Last but not least, various groups have convincingly demonstrated the accessibility of rare architectures like open crystal lattices with fascinating properties through externally addressable and directional interaction potentials.26,27
In the following sections, we will first present approaches towards molecular control over colloidal association followed by their utility for fundamental science and technology. We will present state-of-the-art experimental systems, each with its own limitations and opportunities when evaluated against the desired characteristics described in the above. We hope that this critical review clarifies both the merits and shortcomings of contemporary designs and inspires attempts to reverse-engineer innovative experimental systems that do meet all requirements.
5. Azobenzene-mediated colloidal assembly
Azobenzenes (Fig. 3A) are amongst the most popular photoswitches utilised for a broad range of purposes, including photosurfactants, light-powered motion, photo-switchable surface topologies, and photo-controlled drug release.36–40 Azobenzenes reversibly switch between isomers with and without a dipole moment,41 and thus provide a subtle and elegant approach to manipulate clustering of colloids. In the usually more stable trans isomer, azobenzenes have a dipole moment of 0 debye.41 Upon irradiation with light of a specific wavelength,42 azobenzenes can isomerise into a cis conformation, which for fully hydrogenated azobenzenes has a dipole moment of 4.4 debye.41 When surface-tethered onto particles, the increased dipole moments of the azobenzenes induce dipole–dipole interactions between the particles, which can lead to association of e.g. gold nanoparticles (AuNPs).41–43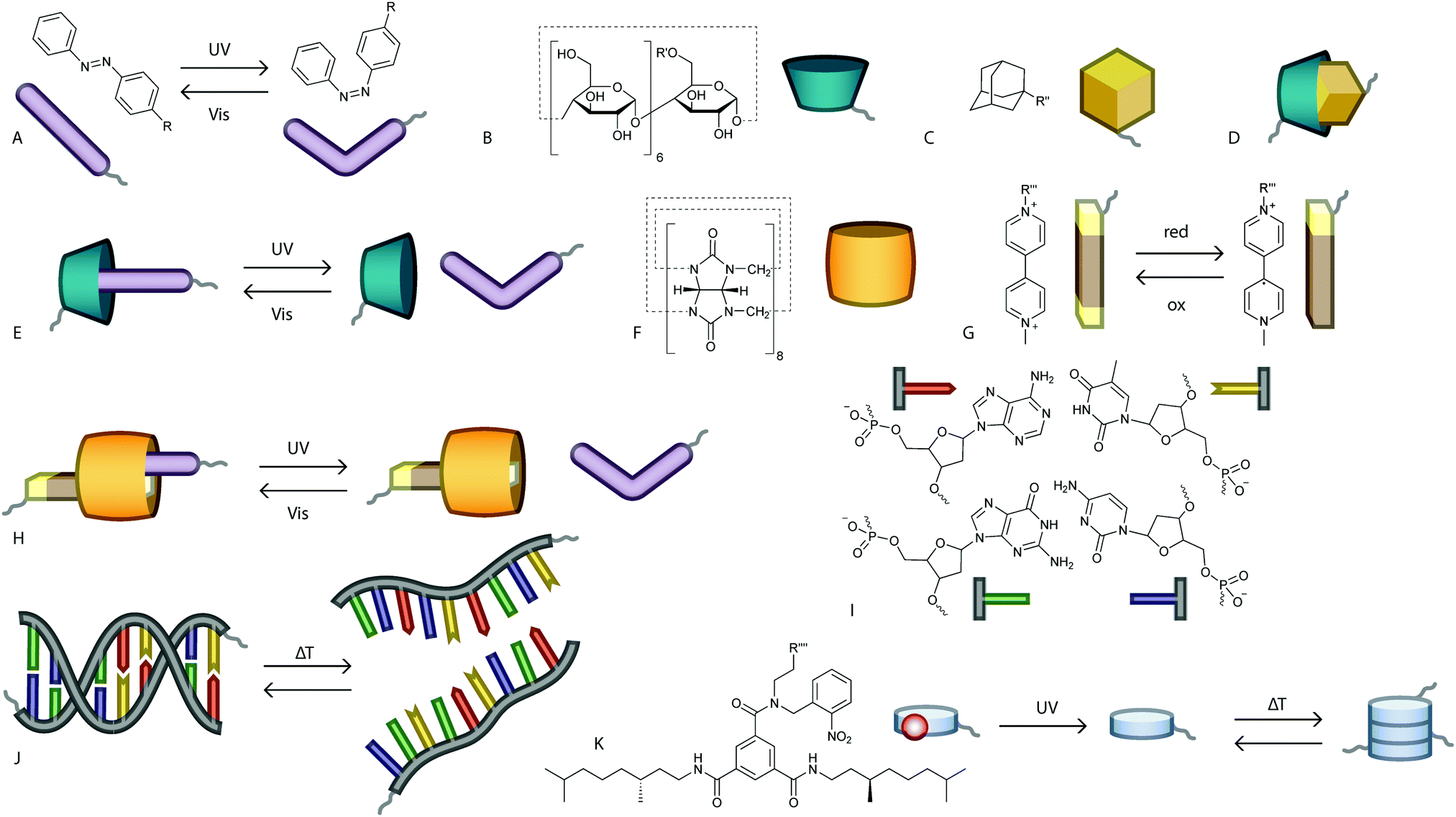 | ||
| Fig. 3 Chemical structures and schematic representations of moieties used in supramolecular colloidal association. (A) trans-Azobenzene can be isomerised to cis-azobenzene upon irradiation with UV light. The reverse process can be induced by irradiation with visible light. Azobenzenes can be attached to the colloidal surface through linker R. (B) β-CD is a ring-like host molecule that is usually tethered to a colloidal surface through a linker (R′) attached to (one of) the methanol group(s). (C) Adamantane is able to form a non-responsive complex with β-CD (D) and can be tethered to the colloidal surface via linker R′′. (E) Azobenzenes and β-CD can form light responsive complexes which can be used to control colloidal assembly. (F) CB[8] is a ring-like host molecule, which remains free in solution until complexation with a suitable surface-immobilised guest. (G) MV2+ is a suitable electron deficient guest for CB[8], can be reduced to disrupt the ternary complex, and can be tethered to the colloidal surface via linker R′′′. (H) A ternary complex of CB[8], MV2+ and azobenzene can be disrupted by UV-light induced isomerisation of the azobenzene. (I) Complementary DNA nucleotides attached to the alternating phosphate and sugar groups that form the DNA backbone. From top left clockwise to bottom left: adenine, thymine, cytosine and guanine. (J) Complementary strands of nucleotides can form temperature responsive duplexes, where the duplex ‘melts’ at high temperatures and re-associates at lower temperatures. (K) O-Nitrobenzyl protected BTAs can be tethered to colloids via linker R′′′′. The photolabile group can be irreversibly cleaved off by irradiation with UV light, after which the BTAs reversibly form temperature responsive assemblies. | ||
In a series of papers, Klajn and co-workers eloquently manipulated the assembly of d = 5.6 nm gold nanoparticles with azobenzene di- and monothiol ligands, which were attached to gold surfaces via well-established chemical methods.41,44 Course-grained calculations showed that the small gold nanoparticles could assemble with as few as 16 azobenzene ligands per particle in cis conformation,41 which was confirmed experimentally by the formation of micrometre-sized crystals from the particles with a low coverage of azobenzene dithiol ligands (∼20 ligands per particle) dispersed in 20–25% v/v methanol in toluene (Fig. 4).41 These reversible crystals remained intact only under continuous UV irradiation, while exposure to visible light or thermal treatment redispersed the gold nanoparticles. Interestingly, an increase in the density of the adsorbed telechelic azobenzene dithiol ligands gave the crystals a permanent character. That is, they remained assembled at high temperatures and under sonication as particles were permanently crosslinked by bridges of ligands that adhered to two particles. As expected, comparable ligands with only a single thiol terminus did not yield such stable ensembles.41–43
 | ||
| Fig. 4 Size control of gold nanoparticle ensembles by irradiation dose. (A) Small ordered structures of only a few nanoparticles are formed after 10 s of irradiation with UV light. Upon further irradiation, free nanoparticles from solution are incorporated in the ‘nuclei’ to form assemblies consisting of hundreds (B and C) to millions (D) of nanoparticles. Scale bars are (A) 5 nm, (B) 20 nm, (C) 50 nm and (D) 200 nm. Copyright 2007 National Academy of Sciences. Ref. 41. | ||
Profiting from the interplay between the optical properties and association of plasmonic particles, rewritable materials were manufactured from AuNPs coated with azobenzene monothiol ligands.43 Light-induced self-assembly (LISA) of AuNPs incorporated into organogel films was triggered locally by application of a photomask during UV-irradiation. The colour of the irradiated regions changed from red to blue as the irradiation dose increased, due to a blue-shift and broadening of the AuNP surface plasmon resonance (SPR) band upon assembly of the metal nanoparticles. Multiple colours can thus be ‘written’ into a film containing only one type of metal particles.
Since UV light is harmful to biological tissues and promotes unwanted side reactions, it is of interest to change the type and position of the substituents of the azobenzene which determine the wavelengths at which the azobenzene isomerises from trans to cis, and vice versa.42,45–47 For example, introduction of a dimethylamino group at the para position allows for trans–cis isomerisation at λ = 420 nm (instead of λ = 365 nm for the parent azobenzene), while irradiation with λ = 365 nm leads to recovery of the trans isomer.42 Klajn and co-workers exploited this feature for selective assembly in mixed populations of nanoparticles functionalised with different azobenzenes, which assembled upon irradiation with either λ = 365 nm or 420 nm. The particles could be redispersed by an increase in temperature, which converted all azobenzenes back into their trans isomers.
Azobenzene-mediated colloidal assembly is clearly versatile as it enables local activation,43 control over the association strength and crystal size,43 the formation of both transient and permanent crystals,41 and light-induced selective assembly.42 However, the impact of remnant, non-negligible van der Waals interactions (due to a mismatch in the refractive indices of the AuNPs and the medium) on these experiments remains largely unclear. Furthermore, the utilised dipole–dipole interactions are not specific interactions, only the independent photo-‘activation’ of different azobenzenes provides selective association of colloids in these studies. Selective interactions can be introduced by utilising other molecular handles such as host–guest complexes.
6. Host–guest mediated colloidal assembly
Selective interactions between a host molecule and one or more guest molecules that are sufficiently small to fit neatly into the cavity of the host are known under the umbrella term host–guest interactions. The formation of such host–guest complexes clearly requires size compatibility and further favourable interactions between the host and guest(s).48 In the case that the guest molecule is too large, it cannot enter the cavity of the host. If the guest molecule is too small, the host–guest interactions may be weaker than if the guest was the right size. In aqueous systems, the cavity of the host is usually hydrophobic in nature, and hydrophobic interactions play an important role in the complex formation. The interplay between the interactions involved in stabilising the complex determines the strength and stability of the formed host–guest complexes. The varying interaction strength for different guests provides an (irreversible) approach to disrupt the initial complexes through addition of competitive guests which interact stronger with the host.β-Cyclodextrin (β-CD, Fig. 3B) is a common ring-like, water-soluble host, often used in combination with the hydrophobic adamantyl (Fig. 3C and D) or trans azobenzene as guests (Fig. 3E).46,49–51 Another frequently used host is cucurbit[8]uril (CB[8], Fig. 3F), which is a ring-like molecule able to form a heteroternary complex with an electron-deficient first guest and an electron-rich second guest.52 Typically, the electron-deficient guest is methyl viologen (MV2+, Fig. 3G) or a derivative thereof.52–55 Naphthol or derivatives thereof are often used as electron-rich guests.52,56Trans azobenzenes are also suitable guest molecules for CB[8], whereas the cis isomer is not (Fig. 3H). It cannot form a complex with CB[8] because of unfavourable steric and electrostatic interactions.57
The selectivity and photoswitchability of host–guest interactions involving azobenzenes have been exploited to control assembly in the colloidal domain. First, a shell of small MV2+-functionalised styrene-based colloids, complexed with CB[8], was immobilised onto an azobenzene-functionalised silica core to generate hybrid raspberry-like colloids (HRCs).58 A densely packed corona could be achieved only for a limited range of number ratios between core and corona particles for a given size ratio. Irradiation with λ = 350 nm induced isomerisation of the azobenzenes and thereby dissociation of the HRCs. The reconstruction of the HRCs upon irradiation with λ = 420 nm was not complete; some of the HRCs aggregated and MV2+-functionalised corona particles remained free in solution.58 Permanent HRCs were prepared via growth of a thin silica layer onto the transient HRCs while in solution. Subsequent heating to sufficiently high temperatures of about 550 °C caused degradation of the styrene particles, leaving behind hollow silica nanospheres enveloping the original silica core.58,59 These may prove useful in, for example, compartmentalised catalysis.
Similarly, hollow mesoporous silica spheres (d = 330 nm) for on-demand cargo release were prepared by first growing a 25 nm thin silica layer onto a polystyrene support, in the presence of worm-like micelles to form the pores, after which the support is dissolved and the silica shell functionalised with azobenzene.60 A corona of MV2+-functionalised iron oxide particles (dTEM = 7 nm) formed on the hollow silica spheres through the formation of a heteroternary complex with CB[8] and turned the hollow particles into containers. Upon irradiation with λ = 350 nm, the azobenzenes isomerised to the cis conformation, disrupted their complexation with CB[8] and thereby released the small iron oxide particles from the surface of the silica spheres. Next, the light-responsive containers were tested for cargo retention. To this end, the hollow silica spheres were loaded with 3-aminophenylboronic acid (BA), which induces a change in colour upon complexation with Alizarin Red salt (AR) which was dissolved in the medium. Thus, the photocontrolled permeability of the nanocontainers could be monitored spectroscopically while the dispersion was subjected to alternating irradiation with λ = 350 nm and 420 nm, causing reversible trans-to-cis and cis-to-trans isomerisation, and as a consequence reversible dissociation and association of the Fe3O4NPs and the silica beads. Strikingly, low molecular weight compounds as small as Mw ∼ 137 g mol−1 were effectively retained as evidenced by the absence of detectable changes in the UV-vis spectra of the dispersing medium after 20 minutes of incubation in the dark (Fig. 5). This compares favourably to other containers, which are reported to be considerably more ‘leaky’, as these lost over 10% of 10 kg mol−1 or larger cargo within 20 minutes.52,56
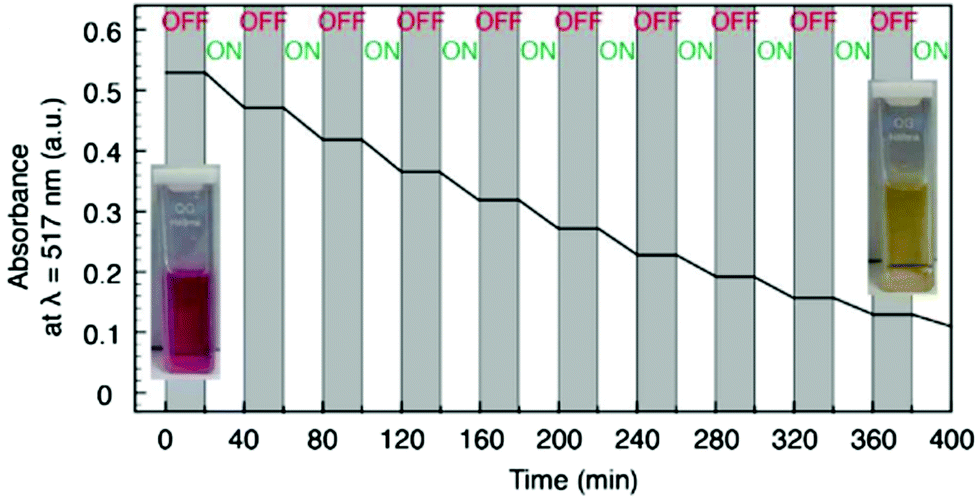 | ||
| Fig. 5 Light-controlled permeability of nanocontainers. The nanocontainers are loaded with 3-aminophenylboronic acid (AB), which forms a complex with Alizarin Red salt (AR) and causes a decrease of the λ = 517 nm absorption peak in the absorption spectrum of the medium. The absorption at λ = 517 nm of the supernatant after centrifugation of the dispersion is plotted against time. The absorption spectrum of the supernatant is recorded every 20 minutes between cycles of irradiation indicated as ‘ON’ (irradiation for 10 minutes with λ = 350 nm, followed by 10 minutes irradiation with λ = 420 nm) and ‘OFF’ (20 minutes incubation in the dark). Inset images show the change in colour of the supernatant upon complexation of AR with BA. Adapted from ref. 60 with permission of The Royal Society of Chemistry. | ||
Larger containers can also be prepared through emulsion templating, in which the container size is set by the size of the emulsion droplet. These range from hundreds of nm's for nanoemulsions up to tens of μm's for macroemulsions, while polymeric supports to produce containers are usually not larger than a few microns in diameter. Particle-stabilised emulsions known as ‘Pickering emulsions’ are an excellent starting point to fabricate robust, hollow supracolloidal structures via crosslinking of the particles once they are adsorbed at the interface. For example, water-in-oil Pickering emulsions coated by AuNPs cross-linked via host–guest complexation can be prepared upon mixing of an aqueous suspension of d = 3 nm AuNPs functionalised with the hydrophilic host β-CD and a dispersion of d = 6 nm AuNPs functionalised with the hydrophobic guest adamantyl in toluene (Fig. 6).50 The large microcapsules of d = 18.3 ± 9.3 μm were found to be stable for several days and tuneable in size through droplet coalescence induced by the addition of adamantyl-functionalised tetra(ethylene glycol) as a competitive guest molecule.
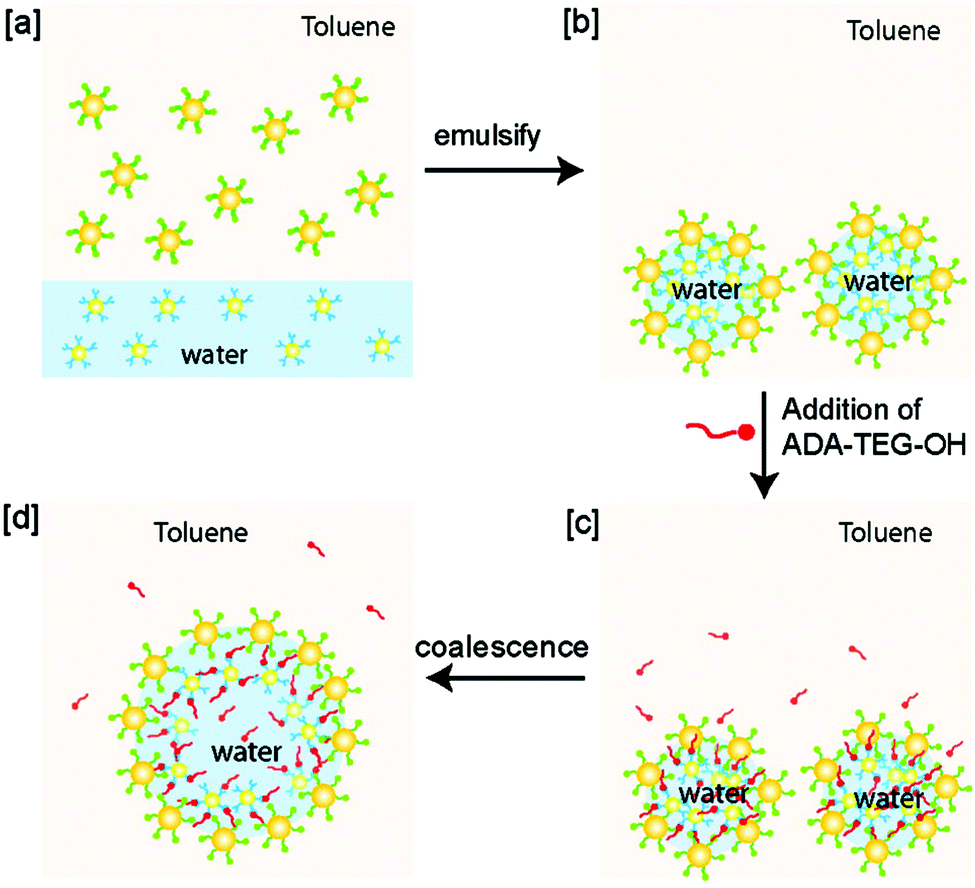 | ||
| Fig. 6 Emulsion templating of microcapsules. (a) Adamantyl-functionalised (green) particles dispersed in toluene, and β-CD functionalised (blue) particles dispersed in water. (b) Emulsification of water and toluene, and host–guest mediated crosslinking of the particles at the interface yields microcapsules. (c) Addition of competitive guest adamantyl-tetra(ethylene glycol)-OH (ADA-TEG-OH, red) disrupts the crosslinking between the particles at the interface, and leads to coalescence (d). Adapted with permission from ref. 50 Copyright 2009 American Chemical Society. | ||
Through careful spatial control over the colloidal surface functionalisation, selective β-CD mediated host–guest interactions can be made directional. A means to this end is the selective functionalisation of isolated areas on patchy particles with either β-CD or its guests. To create particles with suitably sized patches, Huskens and co-workers first covered a monolayer of d = 5 μm silica particles with a layer of spincoated poly(methyl methacrylate) (PMMA), which can be partially removed by O2 plasma etching.25 The size of the exposed silica surface could be tuned by manipulation of the duration of the plasma etching: the longer the etching time, the thicker the PMMA layer that could be removed, thus exposing a larger area on the particle surface. Subsequently, the silica surface could be selectively modified at the exposed area while the particles are still partially embedded in the PMMA, after which the remaining PMMA was gently removed by rinsing with appropriate solvents to enable functionalisation of the newly exposed area. Colloidal assembly into dimers of complementary particles functionalised with either β-CD or adamantyl was demonstrated for colloids with a sufficiently small size of the functionalised patch corresponding to angles below 30° (Fig. 7). The specificity of the interactions could be easily identified through fluorescent colour coding of the functionalised colloids.25 Janus particles with two functionalised patches of a suitable size should give access to more complex architectures like extended (open) lattices.27
 | ||
| Fig. 7 Schematic representation of maximum patch size for dimerization. If the patch has an opening angle of 30° or larger, one particle can in theory interact with multiple other particles. | ||
Using trans azobenzene instead of adamantyl as a guest renders host–guest complexation with β-CD light-responsive. This approach has been implemented for the light-induced reversible association of β-CD- and azobenzene-functionalised silica particles of d ≈ 10 nm into disordered structures upon irradiation with λ = 365 nm.49
Although evidently promising as a molecular tool to guide colloidal assembly, the limited selectivity of host–guest interactions prohibits precise control in multi-component mixtures. To self-organise particles in these materials with high fidelity into single-species (or conversely mixed) clusters requires instead tuneable specific interactions.
7. DNA-mediated colloidal assembly
Hybridisation of single strand deoxyribonucleic acid (ssDNA) with complementary strands has become a most popular means to mediate assembly of particles into both ordered and disordered soft materials with unique optical, mechanical, magnetic, and catalytic properties.61–69 Its high binding specificity originates in the nucleotide sequence along the DNA backbone, as the nucleotides adenine (A) and thymine (T) bind through double hydrogen bonds, while guanine (G) and cytosine (C) form triple hydrogen bonds (Fig. 3I). Various other factors besides nucleotide sequence – including pH, salt concentration and temperature – also impact the thermodynamics of DNA hybridisation (Fig. 3J).70,71 Conveniently, the process is now well understood, which makes it possible to accurately tailor DNA sequences (using free online software tools) to meet certain requirements of e.g. binding specificity and melting temperature, and to subsequently purchase the desired strands from a variety of commercial sources.Pioneering attempts to utilise the binding specificity of DNA to control particle assembly were published in 1996.72,73 Alivisatos et al. functionalised short ssDNA sequences with a single d = 1.4 nm gold nanocrystal, which subsequently hybridised with a complementary ssDNA segment to form dimers or trimers.72 Mirkin et al. made use of a linker, consisting of a short double stranded DNA (dsDNA) segment with two ssDNA ‘sticky ends’. Upon addition of the linker to two mutually non-complementary populations of ssDNA functionalised gold nanoparticles of d = 13 nm, the particles self-assembled into large reversible clusters.73
Roughly ten years later, the first reversible crystalline assemblies of micron-sized colloids were reported.61,71,74–76 This constitutes a grand challenge since kinetic trapping into disordered states en route to thermodynamically favoured crystalline structures is common, due to the strong interaction between ssDNA strands and the large number of DNA-mediated connections between micron-sized colloids.62,71,75 Counterintuitively, a denser DNA coating on the large colloids may (partially) solve this problem.74,77 A sparse and random functionalisation of large colloids can yield bonds that are strong enough to mediate stable clustering, but the low coverage causes an inhomogeneous interaction potential. Increasing the surface density of functional DNA strands and simultaneously weakening their interaction strength creates a soft(er) and more homogeneous attractive field around each colloid.74,77 Similarly, a smooth particle surface generates a more sphero-symmetrical interaction potential than a rough surface.75 Other strategies investigated to circumvent kinetic traps utilise palindromic sequences and mobile linkers.65,78–80 Palindromic sequences are (partially) self-complementary ssDNA sticky ends, which can (partially) self-protect (i.e., self-hybridise), which offers an additional means to control association kinetics, namely through (time dependent) thermal quenching.78,79 Enveloping colloids in a lipid bilayer yields particles with a mobile surface layer into which functional DNA sticky ends can be anchored.65,80 A direct comparison between clustering of colloids coated with mobile and immobile strands shows that lateral freedom of motion of the interparticle links may facilitate crystallisation, as clusters from particles with mobile DNA linkers are more ordered and compact than colloidal clusters from beads carrying immobile strands (Fig. 8).80
 | ||
Fig. 8 Association of colloids with immobile or surface-mobile DNA linkers. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of complementary functionalised particles are cooled from 52 °C, where the particles are fully dissociated, to 25 °C, where the particles are fully associated. (A) Particles of d = 1.0 μm with immobile DNA linkers form disordered structures after quenching the temperature in 15 minutes. (B) Particles of d = 2.0 μm with mobile DNA linkers form more ordered structures upon quick (15 minutes) quenching of the temperature. (C) Upon heating the sample from (B) to 52 °C and quenching to 25 °C over the course of 3 hours, more ordered colloidal clusters are formed. Adapted with permission from ref. 80. Copyright 2013 American Chemical Society. :1 mixtures of complementary functionalised particles are cooled from 52 °C, where the particles are fully dissociated, to 25 °C, where the particles are fully associated. (A) Particles of d = 1.0 μm with immobile DNA linkers form disordered structures after quenching the temperature in 15 minutes. (B) Particles of d = 2.0 μm with mobile DNA linkers form more ordered structures upon quick (15 minutes) quenching of the temperature. (C) Upon heating the sample from (B) to 52 °C and quenching to 25 °C over the course of 3 hours, more ordered colloidal clusters are formed. Adapted with permission from ref. 80. Copyright 2013 American Chemical Society. | ||
Addition of free ssDNA strands to a binary mixture of complementary DNA-functionalised colloids provides an extra handle to control the colloidal phase behaviour through displacement reactions. The competition between enthalpy and entropy allows complex phase behaviour by stabilising dispersions of singlets at low temperature, solid phases at higher temperature, and finally dispersions of singlets above the melting temperature at which all DNA is dissociated.81
DNA-mediated colloidal assembly into a range of lattices that are isostructural with fcc, bbc, hcp, CsCl, Cr3Si, AlB2, and Cs6C60 demonstrates the versatility, the precision and the utility of this unique approach to synthesise reversible supracolloidal structures (Fig. 9).82–85 But, also this strategy has its drawbacks. DNA, like most biological macromolecules, is only stable in a precisely controlled aqueous environment, thus excluding application in organic solvents. This restriction has further implications, as most colloids cannot be refractive index or density matched in water, resulting in a non-specific van der Waals attraction between the particles that must be compensated. In addition, virtually every desired DNA sequence is commercially available, but it is expensive. Finally, DNA-mediated assembly into highly crystalline lattices is slow (on the order of days) and responsive to only temperature as reversible external trigger.68,80,86 It would be interesting to accommodate for additional external triggers, like light, which can be applied with higher spatiotemporal resolution. Recent studies on azobenzene-functionalised oligonucleotides demonstrated the exciting possibility of light-responsive DNA hybridisation87–89 and DNA-mediated colloidal association.90 Since the trans isomer can intercalate in between bases, while the cis isomer cannot, UV-light induced trans-to-cis isomerisation of pendant azobenzenes triggers thermoreversible dehybridisation of azo-dsDNA.
 | ||
| Fig. 9 Crystallisation of DNA-functionalised colloids. Confocal microscopy images of different crystals and crystal planes of DNA-functionalised colloids, and their schematic representations. The colloids are functionalised with palindromic self-complementary ssDNA (‘P’, red), or complementary ‘A’ (green) and ‘B’ (red) ssDNA. The fluorescent dye is incorporated in the surface-functional group. Crystals are assembled from colloids of different sizes and stoichiometries. Scale bars are 5 μm. (a) A face-centred cubic (FCC) lattice is formed from d = 1 μm P particles. The 111, 100, 110 and 311 crystal planes are displayed. (b) d = 1.0 μm A and complementary 1.0 μm B particles form a lattice that is isostructural to CsCl. The 110, 100, 111, and 211 planes are displayed. (c) d = 1.0 μm A and 0.54 μm complementary B particles for a lattice that is isostructural to AlB2. The 100, 001, 111 and 101 planes are displayed. (d) d = 1.5 μm A and 0.54 μm complementary B particles form a lattice that is isostructural to Cs6C60. The 110 and 100 planes are displayed, together with the red channel image showing only the small B particles. ‘Colloidal crystals assembled from DNA-coated colloids’, by Y. Wang et al., ref. 74, is licenced under CC BY 4.0. | ||
8. Hydrogen-bond mediated colloidal assembly
Inspired by hydrogen-bonding motifs in nature, various synthetic compounds have been developed to self-assemble through hydrogen-bonding in a temperature dependent manner in a wide range of solvents. Derivatives of benzene-1,3,5-tricarboxamide (BTA, Fig. 3K), for example, form helical columnar structures as the carboxamides interact though an extensive network of triple hydrogen bonds between adjacent molecules (Fig. 10).91 Interestingly, this supramolecular polymerisation is both temperature and solvent dependent as these affect the strength of the intermolecular hydrogen bonds. Furthermore, molecular association is readily detectable by circular dichroism (CD) spectroscopy provided a stereogenic centre is included in one or more of the BTA side chains.92,93 This incorporation generates a surplus of either left or right handed helices, which can be monitored sensitively by CD spectroscopy. Moreover, hydrogen-bonding can be sterically hindered by a photolabile group, which enables UV-light initiated assembly upon photocleavage of the protecting group from the ‘caged’ BTA.93 In summary, responsive hydrogen-bonding motifs have been engineered for dispersion and reversible assembly in solvents of various polarities,94 which may be exploited as handles for colloidal assembly upon covalent attachment to the surface of particles.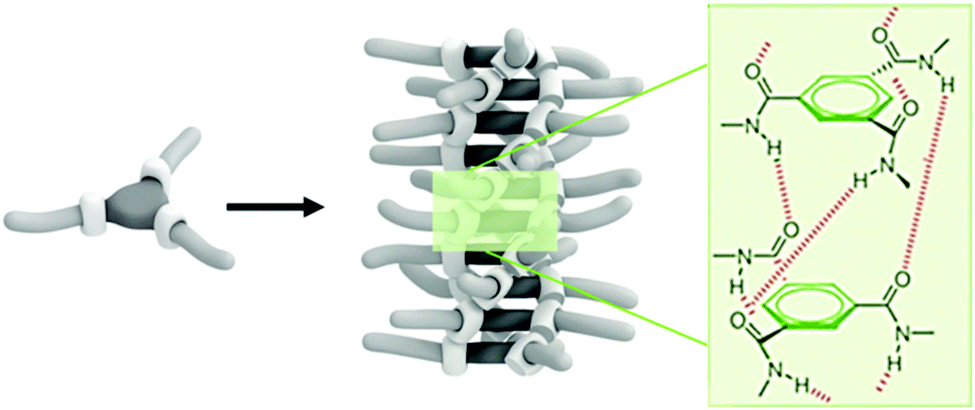 | ||
| Fig. 10 Schematic representation of one dimensional BTA self-assembly. The helical assemblies are stabilised by a triple-helical network of hydrogen bonds, surrounding the core. Adapted from ref. 94 with permission from The Royal Society of Chemistry. | ||
Recently, stearyl alcohol coated silica particles, dispersed in cyclohexane, were covalently functionalised with BTAs ‘caged’ with o-nitrobenzyl and equipped with a stereogenic centre to yield both light and temperature responsive colloids (Fig. 11).93 Confocal microscopy experiments demonstrated that fluorescent particles of d = 513 nm with a 20% BTA surface coverage remained primarily in a singlet state (fsinglet ∼ 0.8) prior to UV irradiation, as the BTAs were ‘inactive’ and the colloids sterically stabilised and refractive index matched by the stearyl alcohol coating. The singlet fraction decreased rapidly upon UV irradiation at λ = 354 nm as colloidal clusters grew in number and size as an increasing amount of tethered BTAs was uncaged and free to interact. Additional light scattering experiments on d = 26 nm functionalised colloids demonstrated thermo-reversible colloidal association and dissociation upon repetitive cycling between 20 °C and 75 °C.93 Interestingly, the time-dependent UV light activation and the broad transition regime between the clustered and dispersed states (∼10 °C) reveal the exciting potential of external control over the multivalency and interaction strength of these so-called supramolecular colloids.
 | ||
| Fig. 11 Light and temperature responsiveness of BTA-functionalised colloids. (a) Schematic representation of cleaving off the photolabile group of surface tethered BTAs by UV light, thereby allowing the BTAs to assemble. (b) Before UV irradiation the green fluorescent colloids are dispersed, and they form clusters upon exposure to UV light. Increasing the temperature disrupts the hydrogen bonds between BTAs and enables redispersion of the colloids, which will cluster again upon lowering of the temperature. Reprinted with permission from ref. 93. Copyright 2015 American Chemical Society. | ||
The above examples show that supramolecular motifs are clearly promising tools to achieve precise control over surface forces. Essential features like solvophilicity, responsivity, external addressability, stability, specificity, tuneability, and detectability are encoded in the molecular design and can be transposed from the molecular to the colloidal domain. However, to fully exploit the power of this approach and create unprecedented, well-ordered three-dimensional structures with emergent properties requires further improvements to the molecular and particle design. Simultaneous refractive index and density matching is one aim, on-off light-switchable (as opposed to light-activatable) interactions (without compromising tuneability in strength in the on-state) is another. Furthermore, non-centrosymmetric particles with shape- or surface anisotropy should be functionalised to profit from the directionality of the supramolecular interactions and finally, the premise of spatiotemporal control over surface forces should be critically tested.
9. Perspective
In the previous sections we have discussed various strategies to guide the assembly of colloids, each with their own limitations and opportunities. For example, non-specific van der Waals, electrostatic, solvophobic or depletion interactions can be combined with a clever design of the colloidal shape or surface properties to yield predictable and complex architectures. However, these interactions are not reversible nor externally addressable. In the experimental systems discussed here, reversibility and external addressability are generally established through introduction of hydrogen-bonding motifs, which are responsive to temperature, or light-switchable azobenzenes, either on their own or in host–guest complexes. We anticipate great advances in the near future when non-spherical and/or patchy particles are site-specifically coated with responsive molecules, in particular with light-switchable hydrogen bonding motifs that associate or interact with high binding specificity. As discussed in the above, key to robust molecular control over colloidal association is accessibility to short inter-particle distances corresponding to the range of weakly attractive intermolecular interactions.11 Let us now look forward and focus on its utility for fundamental science and technology.9.1 From colloids as ‘big’ atoms to colloidal ensembles as ‘big’ molecules and polymers
Profiting from their large size and slow Brownian motion, colloidal particles have been utilised for decades as convenient model systems to better understand the behaviour of atomic materials, which is difficult to probe experimentally as time and length scales are short. Major breakthroughs in colloidal particle synthesis and functionalisation have shifted the focus of this analogy in recent years from utilising spherosymmetrical colloids as models for atomic materials to more complex particle ensembles as models for molecules and (supramolecular) polymers.95–97 Colloidal analogues of polymer chains were created by fixation of strings of colloids formed within AC electrical fields.98 Reversible supracolloidal polymers have been prepared from mixtures of colloidal analogues of ditopic and monotopic monomers and used to test theories developed to describe supramolecular polymerisation.29 Patchy colloids with tailored valence and specific patch–patch interactions have been prepared in a multistep synthesis. An emulsion-evaporation method is used to first produce ‘minimal-moment’ clusters of amidinated polystyrene particles followed by a two-stage swelling process and polymerisation, after which DNA is attached to the ‘bare’ amidinated patches remaining from the original clusters via a biotin–streptavidin–biotin linkage.99Notice that most studies covered in this perspective are performed on colloids of a few (tens of) nanometres. With colloids of this scale, the functional layer is of significant thickness when compared to the particle size and thus can partially ‘correct’ common imperfections like deviations from a perfectly spherical particle shape and interaction potential. By contrast, the surface roughness of micron-sized colloids is often commensurate to the thickness of the functional layer. This may induce inhomogeneities in the interaction potential,75 which reduce control over the interparticle interactions, and must thus be prevented if ordered structures are targeted. Precise engineering of surface roughness, particle shape grafting density, linker length, ligand mobility amongst other features as well as operating conditions (e.g., quench rates) are thus of crucial importance and reliable assembly into ordered architectures is challenging.61,75,100–102 Often, optimisation is done empirically, but experimental and computational studies have identified design principles to avoid kinetic traps and maximise thermodynamic yields of specific (symmetric) target clusters despite strong degeneracy.11,86,103–106 In addition, several studies have demonstrated that fast kinetically guided assembly may lead to interesting finite-sized clusters and bigels, arguing that kinetic traps can also be exploited purposely rather than merely avoided.107–109
We anticipate further advances in this area when progress in particle synthesis, surface functionalisation, assembly pathways, and driven assembly (by e.g. electrical fields, evaporation, capillary action) are combined and further optimised to develop a robust and scalable approach to create complex ordered supracolloidal lattices from anisotropic and patchy particles with externally addressable attractions.100,110–115
9.2 Manufacturing complex colloidal materials
Selective co-organisation of various building blocks in multi-component materials poses challenges that can be met only through purposely engineered complementarity in size, geometry and interactions.85,100,116 DNA-mediated assembly has been studied extensively for this purpose, while the possibilities of molecular control have remained little explored so far. Advantages of the latter approach include synthetic versatility (including modularity in design, high stability, low cost, and high yield) and compatibility with a broad range of solvents as well as other materials like polymers. For example, supramolecular colloids and polymers could be mixed to prepare hybrid organic–inorganic materials harnessing the properties of both elements for enhanced processability, mechanical integrity, transparency, (enantioselective) catalysis, sensing, piezoelectricity, spin filtering, lasing, memory effects, etcetera.117–121 Boyd and co-workers demonstrated that heterogeneously composed, geometrically achiral nanoparticle ensembles can exhibit a chiral optical response, which depends on both the features and arrangement of the constituent particles.122 Molecular control over nanoparticle co-assembly may afford a scalable synthesis route for the studied trimers, which were now fabricated on a substrate by a time-consuming pick-and-place technique.122 When exclusively bottom-up approaches fail, these may be combined with top-down (lithography) or templating methods to prepare complex, multi-component materials for e.g. optoelectronics, multimodal imaging, sensing, and separation technology.123–128 Other application areas include theranostics, spin filter materials, field-effect transistors, data storage devices, and catalysis.129,1309.3 Rational design and synthesis of rare architectures
Although the basic principles of colloidal assembly into clusters, gels, and crystals are known, rational design of colloidal particles aiming to achieve with certainty assembly into a desired architecture remains a formidable challenge. Theory and computer simulations may offer a sense of direction to colloidal chemists focusing on the synthesis and functionalisation of colloidal particles of increasing complexity.131 For example, Whitelam describes assembly into unusual structures, like Archimedian tilings, from patchy particles interacting through complementary patch–patch attractions provided kinetic traps can be avoided.132 Interestingly, both particle geometry and chemical selectively were found essential; i.e., without either of these constraints, Archimedian tilings were not observed. Such particles may become available in the near future when advances in both colloidal particle synthesis and surface functionalisation are exploited and combined into a unified synthetic approach.24,133–135 Illustrative for the potential of this route are elegant examples of DNA-mediated nanoparticle assembly into co-crystals and previously unobserved lattices.100,136 An interesting alternative strategy may be based on 3D DNA origami.137,138 Key here is to separate intraparticle from interparticle DNA hybridisation, which can be realised through inclusion of ‘connector strands’ for interparticle association at opposite faces of DNA cuboids with melting temperatures much lower than those of the strands that hybridise within the cuboids.1379.4 Delivery, sensing, catalysis, diagnostics, and theranostics
Functionalised porous particles and assemblies thereof can be utilised for (gated) cargo uptake, transportation, and on-demand release. For example, drug-loaded mesoporous particles can be coated with (macro)molecular ‘gatekeepers’ to prepare stimuli-responsive nanocontainers through controlled dissociation or (de)swelling of the (macro)molecular valves.139 Alternatively, host–guest interactions have been explored to cap mesoporous silica nanoparticles (MSNs) with AuNPs for light-gated on-demand release of actives from the porous silica containers.140 Using rhodamine B (RhB) as a hydrophilic drug model, Yang and co-workers studied the co-assembly (and triggered dissociation) of pillarene-modified AuNPs and RhB-loaded quaternary ammonium salt functionalised MSNs for this purpose.140 Weinstock and co-workers demonstrated that d = 200 nm supraspheres composed of d = 4 nm AuNPs coated with a mixture of suitable capping agents can uptake unusually large quantities of various organic guests like bisphenol A, azulene, and para-dichlorobenzene from solution, while remaining stable for at least six months at room temperature.141 The hydrophobic voids between the small AuNP within the large supraspheres could accommodate ∼2 million molecules per suprasphere, corresponding to <100 equivalents per d = 4 nm AuNP and less than 43% occupied void space.Colloids and colloidal clusters can serve as nanocontainers for stimuli-controlled uptake, delivery and release, but can also play an active role in chemical reactions, diagnostics, and therapeutics. Photo-isomerisation of tethered azobenzenes can switch ON/OFF the near-infrared (NIR) emission of AuNPs, which may find use in e.g. diagnostics upon further optimisation and transposition from chloroform to physiologically relevant conditions.142 The Klajn group reported acceleration and altered stereoselectivity in ‘self-assembling nanoflasks’ for various chemical reactions in water-saturated toluene upon confinement of small molecules within the ‘interstitial’ spaces of supraparticles assembled from AuNPs coated with light-responsive ligands.143 Qu and co-workers reported enhanced catalytic activity and high enantioselectivity of microbes encapsulated in a W/O Pickering emulsion stabilised by spiropyran-decorated nanoparticles.144 The system could be reversibly inverted from W/O to O/W upon NIR and visible light irradiation.
Another fascinating prospect is further refinement of the mechanical response of shear-activated nanotherapeutics comprising suitably sized aggregates of smaller drug-loaded particles that release their payload when aggregate dissociation is triggered mechanically by e.g. vascular narrowing.145
9.5 Optical and mechanical properties
Guiding colloidal assembly into specific heteroclusters and crystal lattices can yield materials with interesting optical properties which can be exploited for sensing, photonics, diagnostics, and other applications.26,146,147 Analyte-induced clustering of DNA-coated plasmonic nanoparticles is widely studied for the detection of small molecules, metal ions, and nucleic acids.148 Inspired by nature, coatings can be manufactured with structural colours, produced by interference of light arising from structural features within the colloidal material instead of dyes or pigments.149,150 Colloids can enhance the performance of photovoltaics in manifold ways, by affecting scattering, absorption, and near-field enhancement.146 As the nature and arrangement of the (plasmonic) particles dictate the optical features of colloidal materials, we anticipate that molecular control over colloidal (co-)assembly in solution, at interfaces and upon processing into coatings, will help to boost performance. Interesting targets include an increased Q-factor of localised surface plasmon resonance (LSPR)-based devices, tailored iridescence in coatings, and rapid colour-switching for adaptive lasing and sensing of e.g. gases or small molecules.151,152Fine-tuning of the mechanical properties of colloidal materials is another important goal for which molecular control over the range and strength of particle–particle interactions may prove beneficial. As the composition and architecture of a material are both critical determinants of its mechanics, not only the nature of its constituents, but also their assembly pathway and final arrangement matters. Whereas complex architectures of DNA-coated nanoparticles are fragile as a consequence of the low particle volume fraction (∼2–4%), DNA-guided assembly of micron-sized particles affords more robust, densely packed colloidal materials with a much smaller solvent volume fraction.82 The morphology can further be adapted to withstand higher stress, for example, Foffi and co-workers report that bigels can bear higher loads compared to the usual one-component gels.153
9.6 Light-controlled toggling of surface forces
The utility of photoresponsive molecules for light-controlled colloidal assembly has been demonstrated in both organic and aqueous solvents using either surface-tethering to synthesise photoresponsive particles, or instead, direct dissolution to prepare photoresponsive media.93,154 Examples in aqueous media include host–guest mediated formation of silica supraparticles via hydrophobic, surface-grafted azobenzene molecules,49 and dissipative assembly of 6-mercaptohexanoic acid functionalised AuNPs in the presence of water-soluble photoacids.155 These photoacids release protons upon irradiation with visible light, which are recaptured in the dark. These studies exemplify the opportunities of light-gated toggling for (aqueous) colloidal assembly, but require further optimisation to realise their full potential. Particularly interesting in this respect are computational studies demonstrating how the kinetics of supramolecular bonding modifies assembly pathways, without fundamentally altering the nature of the thermodynamically favoured state.156 This means that spatiotemporal ON/OFF toggling of surface forces with a suitably chosen frequency and amplitude (i.e., interaction strength in ON state) aids predictable assembly into architectures that may otherwise remain inaccessible due to the presence of kinetic traps and various competing assembly pathways.10. Concluding remarks
In sum, tailoring the structure and composition of hierarchical soft materials on all length scales spanning from the dimensions of the individual building blocks to the meso- and macroscopic length scales of their assemblies requires precise control over the synthesis of both the building blocks and the materials made thereof. First, particle size, shape and surface chemistry must all be carefully balanced. Second, colloidal assembly must be predictable and adjustable, ideally via spatiotemporal control over surface forces with external cues throughout the entire process of formation of the final material. Kinetic traps must be avoided, assembly pathways adapted, defects repaired (unless irrelevant for the target properties), and surface forces tuned.157 Essential is a profound understanding of the relation between ligand characteristics, assembly kinetics, and phase behaviour,157 which requires direct comparisons between systematic experimental and computational studies focusing on factors like ligand sequestration, the competition between intra-particle loops and inter-particle bridges, the interplay between assembly kinetics and structure formation, and the (molecular) details of the ligand-mediated surface forces (range, strength, multivalency, surface heterogeneity, lateral mobility). Weakly attractive, reversible inter-particle interactions are often – but not always – desirable. At other times, it may be very useful to ‘fixate’ field- or confinement-induced architectures which are not attainable (at sufficiently high yield) through other means.95,158,159 A marriage between supramolecular and colloidal chemistry opens up unprecedented opportunities in this fundamental playground, as illustrated in numerous fascinating reports on colloidal assembly guided by DNA or molecular photo-switches. We anticipate an increasing interest in this active area of research and look forward to many more examples of molecular control over colloidal assembly into functional materials in the coming years.Acknowledgements
The authors acknowledge the Netherlands Organisation for Scientific Research (NWO ECHO-STIP Grant 717.013.005, NWO VIDI Grant 723.014.006) for financial support. The ICMS Animation Studio (Eindhoven University of Technology) is acknowledged for providing the artwork for Fig. 3.References
- J. C. Fröberg, O. J. Rojas and P. M. Claesson, Int. J. Miner. Process., 1999, 56, 1 CrossRef.
- A. Vrij, J. W. Jansen, J. K. G. Dhont, C. Pathmamanoharan, M. M. Kops-Werkhoven and H. M. Fijnaut, Faraday Discuss. Chem. Soc., 1983, 76, 19 RSC.
- N. Vilanova, I. De Feijter and I. K. Voets, J. Visualized Exp., 2016, e53934 Search PubMed.
- E. H. A. de Hoog, PhD thesis, University of Utrecht, 2001.
- R. P. A. Dullens, M. Claesson, D. Derks, A. van Blaaderen and W. K. Kegel, Langmuir, 2003, 19, 5963 CrossRef CAS.
- H. D. Newman and A. Yethiraj, Sci. Rep., 2015, 5, 13572 CrossRef PubMed.
- T. E. Kodger, R. E. Guerra and J. Sprakel, Sci. Rep., 2015, 5, 14635 CrossRef CAS PubMed.
- R. T. Woodward, C. Hight, U. Yildiz, N. Schaeffer, E. M. Valliant, J. R. Jones, M. M. Stevens and J. V. M. Weaver, Soft Matter, 2011, 7, 7560 RSC.
- M. E. Leunissen, C. G. Christova, A.-P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. van Roij and A. van Blaaderen, Nature, 2005, 437, 235 CrossRef CAS PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600 CrossRef CAS PubMed.
- S. Hormoz and M. P. Brenner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5193 CrossRef CAS PubMed.
- S. Asakura and F. Oosawa, J. Chem. Phys., 1954, 22, 1255 CrossRef CAS.
- S. Asakura and F. Oosawa, J. Polym. Sci., 1958, 33, 183 CrossRef CAS.
- H. N. W. Lekkerkerker and R. Tuinier, Colloids and the Depletion Interaction, Springer Netherlands, Dordrecht, 2011, vol. 833 Search PubMed.
- T. H. Zhang, J. Klok, R. Hans Tromp, J. Groenewold and W. K. Kegel, Soft Matter, 2012, 8, 667 RSC.
- W. C. K. Poon, A. D. Pirie and P. N. Pusey, Faraday Discuss., 1995, 101, 65 RSC.
- D. J. Kraft, R. Ni, F. Smallenburg, M. Hermes, K. Yoon, D. A. Weitz, A. van Blaaderen, J. Groenewold, M. Dijkstra and W. K. Kegel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10787 CrossRef CAS PubMed.
- S. Sacanna, W. T. M. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575 CrossRef CAS PubMed.
- S. Badaire, C. Cottin-Bizonne and A. D. Stroock, Langmuir, 2008, 24, 11451 CrossRef CAS PubMed.
- E. Sanz, M. E. Leunissen, A. Fortini, A. van Blaaderen and M. Dijkstra, J. Phys. Chem. B, 2008, 112, 10861 CrossRef CAS PubMed.
- F. M. Bayer, K. Hiltrop and K. Huber, Langmuir, 2010, 26, 13815 CrossRef CAS PubMed.
- K. Sun, R. Kumar, D. E. Falvey and S. R. Raghavan, J. Am. Chem. Soc., 2009, 131, 7135 CrossRef CAS PubMed.
- K. A. Juggernauth, A. E. Gros, N. A. K. Meznarich and B. J. Love, Soft Matter, 2011, 7, 10108 RSC.
- X. Zheng, Y. Wang, Y. Wang, D. J. Pine and M. Weck, Chem. Mater., 2016, 28, 3984 CrossRef.
- X. Y. Ling, I. Y. Phang, C. Acikgoz, M. D. Yilmaz, M. A. Hempenius, G. J. Vancso and J. Huskens, Angew. Chem., Int. Ed., 2009, 48, 7677 CrossRef CAS PubMed.
- X. Mao, Q. Chen and S. Granick, Nat. Mater., 2013, 12, 217 CrossRef CAS PubMed.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381 CrossRef CAS PubMed.
- Y. Wang, A. D. Hollingsworth, S. K. Yang, S. Patel, D. J. Pine and M. Weck, J. Am. Chem. Soc., 2013, 135, 14064 CrossRef CAS PubMed.
- A. Klinkova, H. Therien-Aubin, R. M. Choueiri, M. Rubinstein and E. Kumacheva, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18775 CrossRef CAS PubMed.
- Y. Wang, A. D. Hollingsworth, S. K. Yang, S. Patel, D. J. Pine and M. Weck, J. Am. Chem. Soc., 2013, 135, 14064 CrossRef CAS PubMed.
- R. Klajn, K. J. M. Bishop, M. Fialkowski, M. Paszewski, C. J. Campbell, T. P. Gray and B. A. Grzybowski, Science, 2007, 316, 261 CrossRef CAS PubMed.
- P. B. Shelke, V. D. Nguyen, A. V. Limaye and P. Schall, Adv. Mater., 2013, 25, 1499 CrossRef CAS PubMed.
- I. B. Liu, N. Sharifi-Mood and K. J. Stebe, Philos. Trans. R. Soc., A, 2016, 374, 20150133 CrossRef PubMed.
- A. D. Celiz, T. C. Lee and O. A. Scherman, Adv. Mater., 2009, 21, 3937 CrossRef CAS.
- M. B. Bannwarth, T. Weidner, E. Eidmann, K. Landfester and D. Crespy, Chem. Mater., 2014, 26, 1300 CrossRef CAS.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203 RSC.
- J. Eastoe and A. Vesperinas, Soft Matter, 2005, 1, 338 RSC.
- D. Liu, C. W. M. Bastiaansen, J. M. J. den Toonder and D. J. Broer, Angew. Chem., Int. Ed., 2012, 51, 892 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70 CrossRef CAS PubMed.
- J. Abid, M. Frigoli, R. Pansu, J. Szeftel, J. Zyss, C. Larpent and S. Brasselet, Langmuir, 2011, 27, 7967 CrossRef CAS PubMed.
- R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305 CrossRef CAS PubMed.
- D. Manna, T. Udayabhaskararao, H. Zhao and R. Klajn, Angew. Chem., Int. Ed., 2015, 54, 12394 CrossRef CAS PubMed.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035 CrossRef CAS PubMed.
- M. Brust, D. J. Schiffrin, D. Bethell and C. J. Kiely, Adv. Mater., 1995, 7, 795 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809 RSC.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684 CrossRef CAS PubMed.
- C. B. Rodell, J. E. Mealy and J. A. Burdick, Bioconjugate Chem., 2015, 26, 2279 CrossRef CAS PubMed.
- K. Isenbügel, Y. Gehrke and H. Ritter, Macromol. Chem. Phys., 2012, 213, 227 CrossRef.
- D. Patra, F. Ozdemir, O. R. Miranda, B. Samanta, A. Sanyal and V. M. Rotello, Langmuir, 2009, 25, 13852 CrossRef CAS PubMed.
- Y. Zhou, D. Wang, S. Huang, G. Auernhammer, Y. He, H.-J. Butt and S. Wu, Chem. Commun., 2015, 51, 2725 RSC.
- J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690 CrossRef CAS PubMed.
- S. Jiang, X. Yang, C. Yang, M. Tong, D. Zou and Y. Wu, Tetrahedron Lett., 2013, 54, 1638 CrossRef.
- D. Li, K. Ren, H. Chang, H. Wang, J. Wang, C. Chen and J. Ji, Langmuir, 2013, 29, 14101 CrossRef CAS PubMed.
- C. Stoffelen, J. Voskuhl, P. Jonkheijm and J. Huskens, Angew. Chem., Int. Ed., 2014, 53, 3400 CrossRef CAS PubMed.
- G. Stephenson, R. M. Parker, Y. Lan, Z. Yu, O. A. Scherman and C. Abell, Chem. Commun., 2014, 50, 7048 RSC.
- F. Tian, D. Jiao, F. Biedermann and O. A. Scherman, Nat. Commun., 2012, 3, 1207 CrossRef PubMed.
- Y. Lan, Y. Wu, A. Karas and O. A. Scherman, Angew. Chem., Int. Ed., 2014, 53, 2166 CrossRef CAS PubMed.
- J. D. Peterson, S. Vyazovkin and C. A. Wight, Macromol. Chem. Phys., 2001, 202, 775 CrossRef CAS.
- C. Hu, K. R. West and O. A. Scherman, Nanoscale, 2016, 8, 7840 RSC.
- N. Geerts and E. Eiser, Soft Matter, 2010, 6, 4647 RSC.
- L. Di Michele and E. Eiser, Phys. Chem. Chem. Phys., 2013, 15, 3115 RSC.
- P. E. Theodorakis, N. G. Fytas, G. Kahl and C. Dellago, Condens. Matter Phys., 2015, 18, 22801 CrossRef.
- X. Zhang, R. Wang and G. Xue, Soft Matter, 2015, 11, 1862 RSC.
- S. A. J. van der Meulen, G. Helms and M. Dogterom, J. Phys.: Condens. Matter, 2015, 27, 233101 CrossRef PubMed.
- S. J. Tan, M. J. Campolongo, D. Luo and W. Cheng, Nat. Nanotechnol., 2011, 6, 268 CrossRef CAS PubMed.
- C. Knorowski and A. Travesset, Curr. Opin. Solid State Mater. Sci., 2011, 15, 262 CrossRef CAS.
- E. Auyeung, T. I. N. G. Li, A. J. Senesi, A. L. Schmucker, B. C. Pals, M. O. de la Cruz and C. a. Mirkin, Nature, 2013, 505, 73 CrossRef CAS PubMed.
- M. J. Campolongo, J. S. Kahn, W. L. Cheng, D. Y. Yang, T. Gupton-Campolongo and D. Luo, J. Mater. Chem., 2011, 21, 6113 RSC.
- B. M. Mognetti, M. E. Leunissen and D. Frenkel, Soft Matter, 2012, 8, 2213 RSC.
- M.-P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4225 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, X. Zheng, É. Ducrot, J. S. Yodh, M. Weck and D. J. Pine, Nat. Commun., 2015, 6, 7253 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, X. Zheng, É. Ducrot, M.-G. Lee, G.-R. Yi, M. Weck and D. J. Pine, J. Am. Chem. Soc., 2015, 137, 10760 CAS.
- P. L. Biancaniello, A. J. Kim and J. C. Crocker, Phys. Rev. Lett., 2005, 94, 58302 CrossRef PubMed.
- M. E. Leunissen and D. Frenkel, J. Chem. Phys., 2011, 134, 84702 CrossRef PubMed.
- M. E. Leunissen, R. Dreyfus, F. C. Cheong, D. G. Grier, R. Sha, N. C. Seeman and P. M. Chaikin, Nat. Mater., 2009, 8, 590 CrossRef CAS PubMed.
- Y. Kim, R. J. Macfarlane, M. R. Jones and C. A. Mirkin, Science, 2016, 351, 579 CrossRef CAS PubMed.
- S. A. J. Van Der Meulen and M. E. Leunissen, J. Am. Chem. Soc., 2013, 135, 15129 CrossRef CAS PubMed.
- W. B. Rogers and V. N. Manoharan, Science, 2015, 347, 639 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549 CrossRef CAS PubMed.
- S. Y. Park, A. K. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204 CrossRef CAS PubMed.
- C. Zhang, R. J. Macfarlane, K. L. Young, C. H. J. Choi, L. Hao, E. Auyeung, G. Liu, X. Zhou and C. A. Mirkin, Nat. Mater., 2013, 12, 741 CrossRef CAS PubMed.
- R. T. Scarlett, M. T. Ung, J. C. Crocker and T. Sinno, Soft Matter, 2011, 7, 1912 RSC.
- Y. Nakasone, H. Ooi, Y. Kamiya, H. Asanuma and M. Terazima, J. Am. Chem. Soc., 2016, 138, 9001 CrossRef CAS PubMed.
- E. Sengupta, Y. Yan, X. Wang, K. Munechika and D. S. Ginger, ACS Nano, 2014, 8, 2625 CrossRef CAS PubMed.
- Y. Yan, S. Samai, K. L. Bischoff, J. Zhang and D. S. Ginger, ACS Sens., 2016, 1, 566 CrossRef CAS.
- Y. Yan, J. I. L. Chen and D. S. Ginger, Nano Lett., 2012, 12, 2530–2536 CrossRef CAS PubMed.
- P. J. M. Stals, J. F. Haveman, A. R. A. Palmans and A. P. H. J. Schenning, J. Chem. Educ., 2009, 86, 230 CrossRef CAS.
- L. Brunsveld, A. P. H. J. Schenning, M. A. C. Broeren, H. M. Janssen, J. A. J. M. Vekemans and E. W. Meijer, Chem. Lett., 2000, 292 CrossRef CAS.
- I. de Feijter, L. Albertazzi, A. R. A. Palmans and I. K. Voets, Langmuir, 2015, 31, 57 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC.
- K. L. Heatley, F. Ma and N. Wu, Soft Matter, 2017, 13, 436 RSC.
- W. Poon, Science, 2004, 304, 830 CrossRef CAS PubMed.
- F. Li, D. P. Josephson and A. Stein, Angew. Chem., Int. Ed., 2011, 50, 360 CrossRef CAS PubMed.
- H. R. Vutukuri, A. F. Demirörs, B. Peng, P. D. J. van Oostrum, A. Imhof and A. van Blaaderen, Angew. Chem., Int. Ed., 2012, 51, 11249 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51 CrossRef CAS PubMed.
- M. N. O'Brien, M. R. Jones, B. Lee and C. a. Mirkin, Nat. Mater., 2015, 14, 833 CrossRef PubMed.
- Y. Zou, S. Jayasuriya, C. W. Manke and G. Mao, Langmuir, 2015, 31, 10341 CrossRef CAS PubMed.
- S. C. Glotzer, M. J. Solomon and N. A. Kotov, Am. Inst. Chem. Eng., 2004, 50, 2978 CrossRef CAS.
- W. M. Jacobs, A. Reinhardt and D. Frenkel, J. Chem. Phys., 2015, 142, 21101 CrossRef PubMed.
- T. I. N. G. Li, R. Sknepnek and M. O. De La Cruz, J. Am. Chem. Soc., 2013, 135, 8535 CrossRef CAS PubMed.
- Q. Yu, X. Zhang, Y. Hu, Z. Zhang and R. Wang, ACS Nano, 2016, 10, 7485 CrossRef CAS PubMed.
- R. J. Macfarlane, M. R. Jones, A. J. Senesi, K. L. Young, B. Lee, J. Wu and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 4589 CrossRef CAS PubMed.
- F. M. Hecht and A. R. Bausch, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8577 CrossRef CAS PubMed.
- F. Varrato, L. Di Michele, M. Belushkin, N. Dorsaz, S. H. Nathan, E. Eiser and G. Foffi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19155 CrossRef CAS PubMed.
- M. Grünwald and P. L. Geissler, ACS Nano, 2014, 8, 5891 CrossRef PubMed.
- F. Benyettou, X. Zheng, E. Elacqua, Y. Wang, P. Dalvand, Z. Asfari, J. C. Olsen, D. S. Han, N. Saleh, M. Elhabiri, M. Weck and A. Trabolsi, Langmuir, 2016, 32, 7144 CrossRef CAS PubMed.
- V. Meester, R. W. Verweij, C. Van Der Wel and D. J. Kraft, ACS Nano, 2016, 10, 4322 CrossRef CAS PubMed.
- B. Liu, T. H. Besseling, M. Hermes, A. F. Demirörs, A. Imhof and A. van Blaaderen, Nat. Commun., 2014, 5, 3092 Search PubMed.
- L. Di Michele, F. Varrato, J. Kotar, S. H. Nathan, G. Foffi and E. Eiser, Nat. Commun., 2013, 4, 1 Search PubMed.
- J. Palacci, S. Sacanna, A. P. Steinberg, D. J. Pine and P. M. Chaikin, Science, 2013, 339, 936 CrossRef CAS PubMed.
- S. Sacanna, L. Rossi and D. J. Pine, J. Am. Chem. Soc., 2012, 134, 6112 CrossRef CAS PubMed.
- F. X. Redl, K.-S. Cho, C. B. Murray and S. O'Brien, Nature, 2003, 423, 968 CrossRef CAS PubMed.
- M. Peterca, M. R. Imam, S. D. Hudson, B. E. Partridge, D. Sahoo, P. A. Heiney, M. L. Klein and V. Percec, ACS Nano, 2016, 10, 10480 CrossRef CAS PubMed.
- R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2009, 131, 4233 CrossRef CAS PubMed.
- A. Boal, F. Ilhan, J. DeRouchey, T. Thurn-Albrecht, T. Russell and V. Rotello, Nature, 2000, 404, 746 CrossRef CAS PubMed.
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657 CrossRef CAS.
- M. Bieligmeyer, S. M. Taheri, I. German, C. Boisson, C. Probst, W. Milius, V. Altstädt, J. Breu, H. W. Schmidt, F. D'Agosto and S. Förster, J. Am. Chem. Soc., 2012, 134, 18157 CrossRef CAS PubMed.
- P. Banzer, P. Woźniak, U. Mick, I. De Leon and R. W. Boyd, Nat. Commun., 2016, 7, 13117 CrossRef CAS PubMed.
- M. Zhang, D. J. Magagnosc, I. Liberal, Y. Yu, H. Yun, H. Yang, Y. Wu, J. Guo, W. Chen, Y. J. Shin, A. Stein, J. M. Kikkawa, N. Engheta, D. S. Gianola, C. B. Murray and C. R. Kagan, Nat. Nanotechnol., 2016, 12, 228 CrossRef PubMed.
- J. Xu and Z. Guo, J. Colloid Interface Sci., 2013, 406, 1 CrossRef CAS PubMed.
- M. Pichumani, P. Bagheri, K. M. Poduska, W. González-Viñas and A. Yethiraj, Soft Matter, 2013, 9, 3220 RSC.
- N. Vogel, M. Retsch, C. A. Fustin, A. Del Campo and U. Jonas, Chem. Rev., 2015, 115, 6265 CrossRef CAS PubMed.
- J. Ge and Y. Yin, Angew. Chem., Int. Ed., 2011, 50, 1492 CrossRef CAS PubMed.
- O. Kruglova, P.-J. Demeyer, K. Zhong, Y. Zhou and K. Clays, Soft Matter, 2013, 9, 9072 RSC.
- J.-H. Choi, H. Wang, S. J. Oh, T. Paik, P. Sung, J. Sung, X. Ye, T. Zhao, B. T. Diroll, C. B. Murray and C. R. Kagan, Science, 2016, 352, 205 CrossRef CAS PubMed.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15 CrossRef CAS PubMed.
- A. V. Tkachenko, Phys. Rev. Lett., 2002, 89, 148303 CrossRef PubMed.
- S. Whitelam, Phys. Rev. Lett., 2016, 117, 228003 CrossRef PubMed.
- B. G. P. Van Ravensteijn, M. Kamp, A. Van Blaaderen and W. K. Kegel, Chem. Mater., 2013, 25, 4348 CrossRef CAS.
- S. Sacanna and D. J. Pine, Curr. Opin. Colloid Interface Sci., 2011, 16, 96 CrossRef CAS.
- C. H. J. Evers, J. A. Luiken, P. G. Bolhuis and W. K. Kegel, Nature, 2016, 534, 364 CrossRef PubMed.
- E. Auyeung, J. I. Cutler, R. J. Macfarlane, M. R. Jones, J. Wu, G. Liu, K. Zhang, K. D. Osberg and C. A. Mirkin, Nat. Nanotechnol., 2011, 7, 24 CrossRef PubMed.
- T. Tigges, T. Heuser, R. Tiwari and A. Walther, Nano Lett., 2016, 16, 7870 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311 CrossRef CAS PubMed.
- G.-M. Bao, L. Wang, H.-Q. Yuan, X.-Y. Wang, T.-X. Mei and M.-R. Qu, RSC Adv., 2016, 6, 109453 RSC.
- X. Wang, L.-L. Tan, X. Li, N. Song, Z. Li, J.-N. Hu, Y.-M. Cheng, Y. Wang and Y.-W. Yang, Chem. Commun., 2016, 52, 13775 RSC.
- Y. Wang, O. Zeiri, M. Raula, B. Le Ouay, F. Stellacci and I. A. Weinstock, Nat. Nanotechnol., 2016, 12, 170 CrossRef PubMed.
- S. Bonacchi, A. Cantelli, G. Battistelli, G. Guidetti, M. Calvaresi, J. Manzi, L. Gabrielli, F. Ramadori, A. Gambarin, F. Mancin and M. Montalti, Angew. Chem., Int. Ed., 2016, 55, 11064 CrossRef CAS PubMed.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2015, 11, 82 CrossRef PubMed.
- Z. Chen, L. Zhou, W. Bing, Z. Zhang, Z. Li, J. Ren and X. Qu, J. Am. Chem. Soc., 2014, 136, 7498 CrossRef CAS PubMed.
- N. Korin, M. Kanapathipillai, B. D. Matthews, M. Crescente, A. Brill, T. Mammoto, K. Ghosh, S. Jurek, S. A. Bencherif, D. Bhatta, A. U. Coskun, C. L. Feldman, D. D. Wagner and D. E. Ingber, Science, 2012, 337, 738 CrossRef CAS PubMed.
- M. Karg, T. A. F. König, M. Retsch, C. Stelling, P. M. Reichstein, T. Honold, M. Thelakkat and A. Fery, Mater. Today, 2015, 18, 185 CrossRef CAS.
- Y. Huang and D.-H. Kim, Nanoscale, 2012, 4, 6312 RSC.
- A. Pandya, A. N. Lad, S. P. Singh and R. Shanker, RSC Adv., 2016, 6, 113095 RSC.
- I. Burgess, M. Lončar and J. Aizenberg, J. Mater. Chem. C, 2013, 1, 6075 RSC.
- K.-Y. Tsao, H.-P. Tsai, K.-Y. A. Lin, Y.-X. He and H. Yang, Langmuir, 2016, 32, 12869 CrossRef CAS PubMed.
- G. Sanchez, D. Curiel, I. Ratera, A. Tarraga, J. Veciana and P. Molina, Dalton Trans., 2013, 6318 RSC.
- J.-G. Park, S.-H. Kim, S. Magkiriadou, T. M. Choi, Y.-S. Kim and V. N. Manoharan, Angew. Chem., Int. Ed., 2014, 53, 2899 CrossRef CAS PubMed.
- L. Di Michele, D. Fiocco, F. Varrato, S. Sastry, E. Eiser and G. Foffi, Soft Matter, 2014, 10, 3633 RSC.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646 CrossRef CAS PubMed.
- D. Samanta and R. Klajn, Adv. Opt. Mater., 2016, 4, 1373 CrossRef CAS.
- Z. M. Sherman and J. W. Swan, ACS Nano, 2016, 10, 5260 CrossRef CAS PubMed.
- S. Jan Bachmann, M. Petitzon and B. M. Mognetti, Soft Matter, 2016, 12, 9585 RSC.
- L. Jiang, J. W. J. de Folter, J. Huang, A. P. Philipse, W. K. Kegel and A. V. Petukhov, Angew. Chem., Int. Ed., 2013, 52, 3364 CrossRef CAS PubMed.
- H. R. Vutukuri, J. Stiefelhagen, T. Vissers, A. Imhof and A. Van Blaaderen, Adv. Mater., 2012, 24, 412 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |