
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3R phase of MoS2 and WS2 outperforms the corresponding 2H phase for hydrogen evolution†
Rou Jun
Toh
a,
Zdeněk
Sofer
 b,
Jan
Luxa
b,
David
Sedmidubský
b and
Martin
Pumera
*a
b,
Jan
Luxa
b,
David
Sedmidubský
b and
Martin
Pumera
*a
aDivision of Chemistry & Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: pumera@ntu.edu.sg; Fax: +65 67911961
bDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague, Technicka 5, 166 28 Prague 6, Czech Republic
First published on 27th February 2017
Abstract
Herein, we compare the bulk, 2H and 3R phases of two most prevalent TMD materials: MoS2 and WS2. The 3R phase outperforms its 2H phase counterpart in hydrogen evolution reaction catalysis and is even comparable with the exfoliated, 1T phase in the case of MoS2.
Transition metal dichalcogenides (TMDs) have gained recognition rapidly due to their layered structure, which is similar to that of graphite.1–3 The properties emerging from the anisotropy of TMD materials have been demonstrated to be useful for advanced energy storage and conversion,4 electrochemical catalysis,5,6 and sensing7–9 applications. Of particular interest is their high catalytic activity towards the hydrogen evolution reaction (HER),10–12 which could support efficient and low-cost production of hydrogen without reliance on precious Pt-based catalysts. Such a characteristic points to the importance of understanding the HER catalytic mechanism, and consequently, the synthesis of TMDs with different morphologies aimed at fine-tuning and improving their performances.
The key role played by sulphur-terminated edges of MoS2 as active sites for the HER has been highlighted in recent theoretical studies.13,14 Superior electrochemical and catalytic activity was demonstrated at edge-sites not only for MoS2, but also for other TMD materials like WS2, MoSe2 and WSe2.15,16 Hence, fabrication of TMDs with increased edge-exposed surface with respect to the basal portion16,17 is one of the strategies out of the many proposed to enhance catalytic properties.18 Moreover, it is a well-known fact that the properties of TMD materials in their bulk states vastly differ from their exfoliated counterparts. In order to enhance the catalytic properties of TMD materials, a commonly employed method is thus, splitting the layers of bulk TMD materials into single or few layer sheets via Li intercalation with organolithium compounds.19–21 Another key factor influencing the catalytic properties of TMD materials is their electrical conductivity. A material with high electrical conductivity enables better communication between the electron source and the active edge sites.22
Transition-metal coordination by chalcogens and the stacking sequences of multiple layers of TMD materials can elicit different electronic properties. As illustrated in Fig. 1, a single-layered TMD generally presents either an octahedral or a trigonal prismatic coordination phase. In multi-layered TMDs, a large variety of polymorphic structures arise, as each individual layer can possess any of the two coordination phases. The three commonly found polymorphs are defined as 1T, 2H and 3R, where the digit is indicative of the number of layers in the crystallographic unit cell, and the latter designates the type of symmetry exhibited; T stands for tetragonal (D3d group), H represents hexagonal (D3h group), and R denotes rhombohedral (C53v group). The 1T form displays metallic behaviour, while both 2H and 3R forms exhibit semiconducting behaviour. Each TMD polymorph possesses unique structures and electronic properties from where different catalytic properties can emerge. Hence, it is imperative to make a comparison of different TMD polymorphs, aiming to understand the influence of polymorph structures on the catalytic properties of TMD materials. Previous works have reported a 2H → 1T phase transition when TMD materials are subjected to chemical exfoliation23 and a comparison of the catalytic properties of 1T- and 2H-TMDs has shown that the exfoliated, metallic 1T phase performs as better catalysts than their bulk, semiconducting 2H counterparts for HER.24 This was explained by the increased edge-exposed surface, reduced sheet size and enhanced electrical conductivity of the 1T phase. However, there has been no previous study and there is a lack of understanding of the catalytic properties of 3R-TMDs or comparison with their semiconducting 2H counterparts.
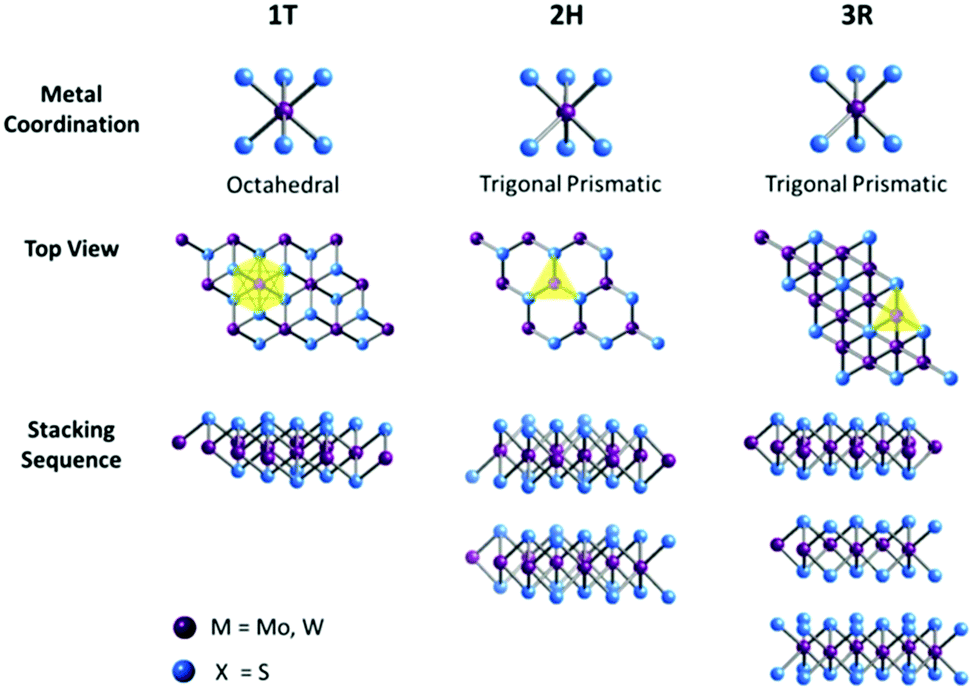 | ||
| Fig. 1 Metal coordinations and stacking sequences of TMD structural unit cells. Metal coordination can be either octahedral or trigonal prismatic. The octahedral coordination allows stacking sequences, which yield a tetragonal symmetry (1T). Dissimilar stacking sequences of trigonal prismatic single layers can give rise to different symmetries: hexagonal symmetry (2H) and rhombohedral symmetry (3R). | ||
In this work, we wish to address the following basic questions: (1) is the catalytic activity towards HER influenced by the polymorph structure and if so, (2) is an additional chemical exfoliation step necessary for producing an efficient HER catalyst. For this purpose, we synthesized TMD materials (MoS2 and WS2) in their 3R phase, which allows us to perform a comparison of their HER catalytic activity with their corresponding 2H phase. The synthesis was performed from oxides using an alkali metal carbonate/sulphur melt. Based on X-ray diffraction (XRD), high resolution transmission electron microscopy (HR-TEM), scanning electron microscopy (SEM), laser diffraction, energy dispersive X-ray spectroscopy (EDS), X-ray fluorescence (XRF), X-ray photoelectron spectroscopy (XPS) and electrochemical analyses, we show that the 3R phase outperforms the 2H phase in HER catalysis. However, this improvement in the HER catalytic property presents in varying magnitudes for MoS2 and WS2, and at different synthesis temperatures. Comparison with existing literature shows that in the case of MoS2, catalytic performance towards HER for the bulk, semiconducting 3R phase is on par with the exfoliated, metallic 1T phase.
Considering the two highly studied transition metal dichalcogenides (TMDs), i.e. MoS2 and WS2, we first synthesized the 3R phase from their respective oxides with sulphur in carbonate flux according to previously established protocols at 550 °C.25,26 Herein, they are denoted as 3R-MoS2 (550 °C) and 3R-WS2 (550 °C). We studied their HER catalytic properties, and further investigated if the polymorph structure influences the HER catalytic performances of MoS2 and WS2 by making a comparison of the 3R phase TMDs with their corresponding 2H phases; 2H-MoS2 (AA) or 2H-WS2 (AA). The materials were fully characterised by using XRD, HR-TEM, SEM, laser diffraction, EDS, XRF, XPS and electrochemically tested as catalysts for hydrogen evolution.
The phase purity of each material was verified by X-ray diffraction (XRD). Fig. S1 (ESI†) represents the XRD patterns of the TMD materials. The 2H-MoS2 (AA) (Fig. S1A, ESI†) and 2H-WS2 (AA) (Fig. S1B, ESI†) were found as a single 2H phase and exhibiting a hexagonal structure, while 3R-MoS2 (550 °C) (Fig. S1C, ESI†) and 3R-WS2 (550 °C) (Fig. S1D, ESI†) were formed as a mixture of the rhombohedral 3R phase and the hexagonal 2H phase. The ratio evaluated from XRD gives a composition of about 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 using the Rietveld refinement method. More details are given in the ESI† (Fig. S2). The presence of the desired 3R stacking of the atomic layers in 3R-MoS2 (550 °C) and 3R-WS2 (550 °C) was further confirmed by HR-TEM analyses in Fig. S3 and S4 (ESI†), respectively. Further characterisation of the materials was performed using SEM (Fig. S5, ESI†), which indicated the homogeneous formation of platelet structures, and laser diffraction measurements revealed comparable particle sizes of the TMD materials in their 2H phase and the respective 3R phase (Fig. S6, ESI†). 2H-MoS2 (AA) and 3R-MoS2 (550 °C) have mean particle diameters of 24.2 μm and 12.9 μm, respectively, while 2H-WS2 (AA) and 3R-WS2 (550 °C) exhibited mean particle diameters of 11.1 μm and 10.4 μm respectively. Moreover, the composition of each material was confirmed via EDS (Fig. S7–S10, ESI†) and XRF (Table S1, ESI†). The concentration of potassium originating from the potassium carbonate flux was 1.2 wt% and 1.0 wt% in 3R-MoS2 (550 °C) and 3R-WS2 (550 °C), respectively. In 3R-MoS2 (550 °C), the concentration of tungsten lies below 0.01 wt%. However, in 3R-WS2 (550 °C), the concentration of molybdenum is 0.15 wt%. In addition, traces of Zn were detected in 3R-MoS2 (550 °C) (∼0.01 wt%), and traces of Fe were found in 3R-WS2 (550 °C) (∼0.02 wt%). The concentrations of all other impurities were below 0.01 wt%. Overall, it was determined that the presence of impurities is insignificant in these samples and would not interfere with the electrochemical signals of the TMD materials.
:1 using the Rietveld refinement method. More details are given in the ESI† (Fig. S2). The presence of the desired 3R stacking of the atomic layers in 3R-MoS2 (550 °C) and 3R-WS2 (550 °C) was further confirmed by HR-TEM analyses in Fig. S3 and S4 (ESI†), respectively. Further characterisation of the materials was performed using SEM (Fig. S5, ESI†), which indicated the homogeneous formation of platelet structures, and laser diffraction measurements revealed comparable particle sizes of the TMD materials in their 2H phase and the respective 3R phase (Fig. S6, ESI†). 2H-MoS2 (AA) and 3R-MoS2 (550 °C) have mean particle diameters of 24.2 μm and 12.9 μm, respectively, while 2H-WS2 (AA) and 3R-WS2 (550 °C) exhibited mean particle diameters of 11.1 μm and 10.4 μm respectively. Moreover, the composition of each material was confirmed via EDS (Fig. S7–S10, ESI†) and XRF (Table S1, ESI†). The concentration of potassium originating from the potassium carbonate flux was 1.2 wt% and 1.0 wt% in 3R-MoS2 (550 °C) and 3R-WS2 (550 °C), respectively. In 3R-MoS2 (550 °C), the concentration of tungsten lies below 0.01 wt%. However, in 3R-WS2 (550 °C), the concentration of molybdenum is 0.15 wt%. In addition, traces of Zn were detected in 3R-MoS2 (550 °C) (∼0.01 wt%), and traces of Fe were found in 3R-WS2 (550 °C) (∼0.02 wt%). The concentrations of all other impurities were below 0.01 wt%. Overall, it was determined that the presence of impurities is insignificant in these samples and would not interfere with the electrochemical signals of the TMD materials.
Moving on with investigating the HER efficiency of the TMD materials, linear sweep voltammetry (LSV) was performed in 0.5 M sulphuric acid at a low scan rate of 2 mV s−1. We studied the HER catalytic performances of the 3R phase and compared them to that of the corresponding 2H phase.
The polarization curves and Tafel plots of 3R-MoS2 (550 °C) and 2H-MoS2 (AA) are represented in Fig. 2A and B, and those of 3R-WS2 (550 °C) and 2H-WS2 (AA) and are shown in Fig. 2C and D. Bare glassy carbon (GC) was included as a material of reference. We adopted three commonly used HER performance indices, namely the overpotential, onset potential and Tafel slope. Overpotentials were taken at −10 mA cm−2, and the onset potential is defined as the potential at which the current density is −0.07 mA cm−2. In general, lower overpotentials and less negative onset potentials indicate higher HER catalytic efficiencies, as less energy is required for the reaction to take place. An analysis of the Tafel slope gives us an idea of the mechanism of the hydrogen evolution on the different TMD surfaces, where the rate-determining step may be determined.22
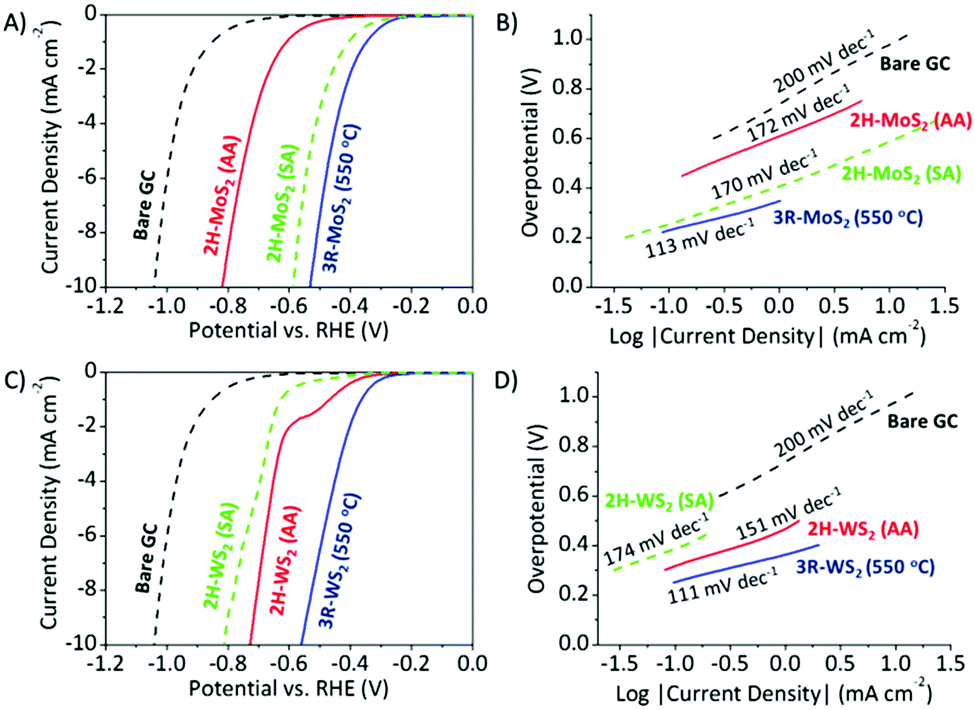 | ||
| Fig. 2 HER polarization curves of 2H and 3R phase TMDs. Linear sweep voltammograms for HER in acidic electrolyte on (A) MoS2 and (C) WS2 compounds. Tafel plots for (B) MoS2 and (D) WS2 compounds. Conditions: background electrolyte, H2SO4 (0.5 M); scan rate, 2 mV s−1. All measurements were performed relative to the Ag/AgCl reference electrode and are corrected to reversible hydrogen electrode (RHE) potentials. | ||
We observed similar and outstanding HER catalytic performances for both materials in the 3R phase. 3R-MoS2 (550 °C) displayed an overpotential of 0.52 V vs. RHE, onset potential of −0.20 V vs. RHE, while 3R-WS2 (550 °C) showed an overpotential of 0.56 V vs. RHE, onset potential of −0.23 V vs. RHE. Tafel analysis of 3R-MoS2 (550 °C) (113 mV dec−1) and 3R-WS2 (550 °C) (111 mV dec−1), which is close to the 120 mV dec−1 theoretical value, generally points to the Volmer adsorption as the limiting step. When compared with 2H-MoS2 (AA), 3R-MoS2 (550 °C) exhibited an overpotential, which was a significant 0.29 V lower. The relatively more negative onset potential of 2H-MoS2 (AA) (−0.41 V vs. RHE) further exemplifies its poorer HER catalytic performance. In the case of WS2, the 2H phase also displayed a poorer HER catalytic performance than the 3R phase, even though to a lesser extent. 2H-WS2 (AA) displayed an overpotential (0.73 V vs. RHE), which is 0.17 mV less than that of 3R-WS2 (550 °C), and a more negative onset potential of −0.30 V vs. RHE. In general, the 2H phase has poorer HER efficiencies than the 3R phase for MoS2 and WS2, with the Tafel slopes of 2H-MoS2 (AA) (172 mV dec−1) and 2H-WS2 (AA) (151 mV dec−1) similarly directing us to the Volmer adsorption as the HER rate-determining step. Moreover, a second TMD material in the 2H phase with different particle sizes was included as an additional control for both MoS2 and WS2. The additional materials are denoted as 2H-MoS2 (SA) and 2H-WS2 (SA), respectively. 2H-MoS2 (SA) and 2H-WS2 (SA) both exhibit a single hexagonal 2H phase (Fig. S11, ESI†) and their particle sizes are 1.8 μm and 4.1 μm, respectively (Fig. S12, ESI†). 2H-MoS2 (SA) displayed Volmer adsorption as the HER rate-determining step, an overpotential of 0.56 V vs. RHE and an onset potential of −0.22 V vs. RHE. Despite having particle sizes ∼7 fold smaller, 2H-MoS2 (SA) showed an inferior HER catalytic performance compared to 3R-MoS2 (550 °C). A similar observation is made with WS2. The additional control experiment further confirms the notion that the 3R phase outperforms its corresponding 2H phase for hydrogen evolution.
The stellar HER efficiency of the 3R phase prompted us to make a comparison with the exfoliated, 1T phase, studied previously by our group.6,24 1T-MoS2 and 1T-WS2 were prepared by the exfoliation of 2H-MoS2 (SA) and 2H-WS2 (SA), respectively which would result in particle sizes of the 1T phase TMDs being similar to or slightly smaller than their 2H counterparts. While 3R-MoS2 (550 °C) exhibited an overpotential of 0.52 V vs. RHE and a Tafel slope of 113 mV dec−1, 1T-MoS2 was reported to display a similar overpotential of about 0.55 V vs. RHE and a Tafel slope of 99 mV dec−1. Despite being in its bulk form and with bigger particle sizes, it was surprising to note that the HER catalytic performance of 3R-MoS2 (550 °C) is comparable to that of the exfoliated, 1T-MoS2. On the contrary, 3R-WS2 (550 °C) did not perform as good as the exfoliated, 1T-WS2; 1T-WS2 reported an overpotential of 0.35 V vs. RHE and a Tafel slope of 85 mV dec−1.
In an attempt to optimise the synthesis conditions of 3R phase MoS2 and WS2 for HER applications, the study was further extended to include two other synthesis temperatures; 650 °C and 750 °C. A comparison of the HER catalytic performances of the 3R phases synthesized at different temperatures was performed, and their polarization curves and Tafel plots are represented in Fig. S13 (ESI†). A careful analysis of their HER performance indicators (Fig. S14, ESI†) disclosed a general trend, applicable for both MoS2 and WS2 materials. In both cases of MoS2 and WS2, the 3R phase displayed superior or comparable HER performance to the 2H phase regardless of synthesis temperatures. However, as the synthesis temperature for the 3R phase is increased from 550 °C to 750 °C, we observed a decreasing trend in the HER efficiency. Hence, we report that the optimal temperature for the synthesis of 3R phase MoS2 and WS2 for HER applications is 550 °C.
Delving deeper to understand the trend observed with different synthesis temperatures for the 3R phase MoS2 and WS2, we consider four possible factors that may affect the HER efficiency of the materials; (1) 3R phase concentration, (2) crystallite size, (3) presence of impurities and (4) particle size. For this purpose, characterisation of the materials was carried out using XPS, XRD, EDS, XRF, SEM and laser diffraction. Fig. S15 (ESI†) shows the high resolution XPS spectra of the 3R phase MoS2 and WS2 synthesized at 550 °C, 650 °C and 750 °C. Deconvolution analyses of the Mo bonding modes show the presence of 2H and/or 3R phases in the 3R-MoS2 (550 °C), 3R-MoS2 (650 °C) and 3R-MoS2 (750 °C) materials with Mo 3d5/2 and Mo 3d3/2 at binding energies of ∼229.0 and 232.5 eV, respectively. Similarly, deconvolution analyses of the W bonding modes show the presence of the 2H and/or 3R phases in the 3R-WS2 (550 °C), 3R-WS2 (650 °C) and 3R-WS2 (750 °C) materials with W 4f7/2 and Mo 4f5/2 at binding energies of ∼33.2 and 35.3 eV, respectively. For comparison, the XPS spectra of 2H-MoS2 and 2H-WS2 measured on single crystals are also shown (Fig. S16, ESI†). In order to distinguish between the 2H and 3R phases, X-ray diffraction was employed. The X-ray diffractograms of the 3R phase MoS2 and WS2 synthesized at 550 °C, 650 °C and 750 °C are shown in Fig. S17 and S18 (ESI†), revealing that the 3R phase was successfully produced at all three temperatures. However, we observed that an increase in synthesis temperature generally led to a decrease in the 3R phase concentration. This is most significant for MoS2, where the concentration of the 3R phase was only 30 wt% for the material synthesized at 750 °C. On the other hand, the concentration of 3R-WS2 was still about 45 wt% for the material synthesized at 750 °C. Moreover, decreasing FWHM of the reflection indicates improvement of crystallinity and increase of crystallite size with increasing synthesis temperature. Therefore, a reduction in the 3R phase concentration and increase in the crystallite size account for the decreasing trend in the HER catalytic performance of the 3R phase MoS2 and WS2 with increasing synthesis temperature.
Further characterization was performed with EDS (Fig. S19–S22, ESI†), which presented no obvious changes in elemental composition regardless of the synthesis temperature. More in-depth analyses with XRF (Table S1, ESI†) reported that the concentration of potassium originating from potassium carbonate flux ranges from 0.05 to 1.2 wt% and decreases with increasing synthesis temperature. While only traces of tungsten were detected in 3R-MoS2, the concentration of molybdenum ranges from 0.15 to 0.39 wt% in 3R-WS2 and this concentration increases with synthesis temperature. Furthermore, similar traces of Zn and traces of Fe were found in 3R-MoS2 and 3R-WS2, respectively, regardless of synthesis temperature. The concentrations of all other impurities were below 0.01 wt%. It can be concluded that the changes in the elemental composition of 3R-MoS2 and 3R-WS2 synthesized at different temperatures were not significant enough to interfere with the electrochemical signals. Next, to study the relation between the particle size and the HER catalytic activity, we performed SEM (Fig. S5, ESI†) and laser diffraction measurements for particle size analysis (Fig. S6, ESI†). All samples are highly polydispersed with particle sizes in the range of 0.2–50 μm. However, a closer observation of the particle size distribution curves from laser diffraction measurements does not reveal any correlation with the synthesis temperature. From these results, we can conclude that the main influence on electrocatalytic activity will be related to the concentration of the 3R phase in the TMD materials and the size of individual crystallites.
A comprehensive study and comparison of the 3R and 2H phases of the two most representative TMD materials, MoS2 and WS2, has been conducted in relation to their catalytic properties towards the hydrogen evolution reaction. We report the synthesis of 3R-MoS2 and 3R-WS2 materials via a simple and scalable method at three different temperatures (550 °C, 650 °C and 750 °C). The optimal synthesis temperature was determined to be 550 °C for applications in industrially important electrocatalysis like HER, where high 3R phase concentration and small crystallite sizes were obtained. It was demonstrated that the 3R phase displayed superior HER efficiency compared to the 2H phase. Additionally, a more significant improvement in the HER catalytic activity was observed with 3R-MoS2 than with 3R-WS2. Specifically, it is surprising to note that the bulk, 3R-MoS2 (550 °C) displayed comparable HER efficiency to that of the previously studied exfoliated, 1T-MoS2. These results highlight the profound impact different polymorph structures of TMD materials have on their HER efficiencies, and further challenge the need for the commonly employed technique of exfoliation to enhance the catalytic properties of TMD materials. The insights from this work not only provide an understanding of the underlying electrochemical activity of TMD materials, but are also particularly important in the search for highly efficient catalysts for hydrogen evolution.
M. P. acknowledges a Tier 2 grant (MOE2013-T2-1-056). R. J. T. acknowledges SMA3 Singapore-MIT SMART Center. Z. S., D. S. and J. L. were supported by the Czech Science Foundation (GACR No. 16-05167S) and by specific university research (MSMT No. 20-SVV/2017). This work was supported by the project Advanced Functional Nanorobots (reg. No. CZ.02.1.01/0.0/0.0/15_003/0000444 financed by the EFRR).
Notes and references
- X. Chia, A. Y. S. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Chem. Rev., 2015, 115, 11941 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766 CrossRef CAS PubMed.
- X. Cao, C. Tan, X. Zhang, W. Zhao and H. Zhang, Adv. Mater., 2016, 28, 6167 CrossRef CAS PubMed.
- S. S. Chou, N. Sai, P. Lu, E. N. Coker, S. Liu, K. Artyushkvoa, T. S. Luk, B. Kaehr and C. J. Brinker, Nat. Commun., 2015, 6, 8311 CrossRef CAS PubMed.
- X. Chia, A. Ambrosi, Z. Sofer, J. Luxa and M. Pumera, ACS Nano, 2015, 9, 5164 CrossRef CAS PubMed.
- A. H. Loo, A. Bonanni, Z. Sofer and M. Pumera, ChemPhysChem, 2015, 16, 2304 CrossRef CAS PubMed.
- K. Kalantar-zadeh, J. Z. Ou, T. Daeneke, M. S. Strano, M. Pumera and S. L. Gras, Adv. Funct. Mater., 2015, 25, 5086 CrossRef CAS.
- D. Sarkar, X. Xie, J. Kang, H. Zhang, W. Liu, J. Navarrete, M. Moskovits and K. Banerjee, Nano Lett., 2015, 15, 2852 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhang, Z. Yang, Y. Yan and K. Sun, Sci. Technol. Adv. Mater., 2013, 14, 043501 CrossRef PubMed.
- J. D. Benck, T. R. Hellsternn, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957 CrossRef CAS.
- (a) Z. He and W. Que, Appl. Mater. Today, 2016, 3, 23 CrossRef; (b) F. Reale, K. Sharda and C. Mattevi, Appl. Mater. Today, 2016, 3, 11 CrossRef.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielson, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Norskov, Phys. Chem. Chem. Phys., 2014, 16, 13156 RSC.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341 CrossRef CAS PubMed.
- D. Y. Chung, S. K. Park, Y. H. Chung, S. H. Yu, D. H. Lim, N. Jung, H. C. Ham, H. Y. Park, Y. Piao, S. J. Yoo and Y. E. Sung, Nanoscale, 2014, 6, 2131 RSC.
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197 CrossRef CAS PubMed.
- M. Kertesz and R. Hoffman, J. Am. Chem. Soc., 1984, 106, 3453 CrossRef CAS.
- W. M. R. Divigalpitiya, S. R. Morrison and R. F. Frindt, Thin Solid Films, 1990, 186, 177 CrossRef CAS.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. W. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chowalla, Nano Lett., 2013, 13, 6222 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed.
- M. A. Py and R. R. Haering, Can. J. Phys., 1983, 61, 76 CrossRef CAS.
- A. Ambrosi, Z. Sofer and M. Pumera, Chem. Commun., 2015, 51, 8450 RSC.
- R. E. Bell and R. E. Herfert, J. Am. Chem. Soc., 1957, 79, 3351 CrossRef CAS.
- J. C. Wilderyanck and R. Jellinek, Z. Anorg. Allg. Chem., 1964, 328, 309 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc09952a |
| This journal is © The Royal Society of Chemistry 2017 |