
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical probing of thiotetronate bio-assembly†
Judith
Havemann
a,
Marie E.
Yurkovich
b,
Robert
Jenkins
a,
Sophia
Harringer
a,
Weixin
Tao
c,
Shishi
Wen
c,
Yuhui
Sun
c,
Peter F.
Leadlay
b and
Manuela
Tosin
 *a
*a
aDepartment of Chemistry, University of Warwick, Library Road, CV4 7AL, UK. E-mail: M.Tosin@warwick.ac.uk; Tel: +44(0) 2476572878
bDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK
cKey Laboratory of Combinatorial Biosynthesis and Drug Discovery (Wuhan University), Ministry of Education, Wuhan University School of Pharmaceutical Sciences, Wuhan 430071, People's Republic of China
First published on 25th January 2017
Abstract
Chemical ‘chain termination’ probes were utilised for the investigation of thiotetronate antibiotic biosynthesis in the filamentous bacteria Lentzea sp. and Streptomyces thiolactonus NRRL 15439. The use of these tools led to the capture of biosynthetic intermediates involved in the thiotetronate polyketide backbone assembly, providing first insights into substrate specificity and in vivo intermediate processing by unusual iterative synthases.
Polyketide natural products constitute a prominent class of secondary metabolites that interact with a wide range of intracellular targets associated with diseases.1 The presence of heterocyclic moieties in polyketides, such as pyran and furan rings in ionophore antibiotics, is common and often decisive for the physical properties and biological activity of the compound.2 Although sulphur-ring based structures are relatively common in peptides of both ribosomal and nonribosomal origin,3 polyketides bearing sulphur-containing rings are rare. They include potent antitumor agents such as leinamycin4 and the family of thiotetronate antibiotics (Fig. 1).5–10 Thiotetronates are characterised by thiolactone moieties functionalised with different substituents at positions 3, 5 and 7, a tetronate moiety spanning carbons 2–4 and a stereogenic centre at carbon 5. The best-known thiotetronate is thiolactomycin (1, TLM): originally isolated from a soil Nocardia strain (ATCC 31319,7 recently reclassified as Lentzea sp.), 1 is a reversible inhibitor of the β-ketoacyl-acyl carrier protein synthase (KAS) enzymes of the bacterial type II (dissociated) fatty acid synthase.11 TLM constitutes a promising lead structure for the development of novel anti-tuberculosis, anti-malarial and anti-trypanosomal agents.12–14 The closely structurally related Tü 3010 (2) has been reported to be the most potent antibacterial tetronate to date, capable of targeting FabH/FabF in Staphylococcus aureus at lower doses in comparison to TLM and other known FabH/FabF inhibitors.15 Only very recently these medicinally promising molecules were unequivocally established to be of polyketide origin.5–6,16 Comparative genomics-based approaches to orphan cluster identification led to the uncovering of putative polyketide synthase (PKS)-nonribosomal peptide synthetase (NRPS) enzymes (A–C, Fig. 1B) responsible for thiotetronate production in both soil and marine bacteria, in association with tailoring enzymes (e.g. P450 oxidases, D), cysteine desulfurases (J) and (thiouridylase-like) sulfur transferases (S) proposed to provide the sulfur atom for thiotetronate ring formation.5,6 Indeed, genes A, B, D1, D2, J and S are essential for thiotetronate formation, as proved by in vivo knock-out and complementation studies.5 Genes A and B encode for an acetyl-loading PKS module and a hybrid PKS-NRPS, respectively. For the thiolactomycin biosynthetic gene cluster tlm, the B protein comprises ketosynthase (KS), acyltransferase (AT), dehydratase (DH), ketoreductase (KR), acyl carrier protein (ACP), cyclisation (Cy), adenylation (A), peptidyl carrier protein (PCP) and thioesterase (TE) domains (Fig. 3). A number of biosynthetic hypotheses have been so far proposed regarding the origins of TLM and other thiotetronates on the basis of precursor feeding studies16 and gene/protein bioinformatics.5,6 However, the actual molecular mechanisms of product assembly remain undetermined and are the object of intense scrutiny with a view to thiotetronate structural diversification via enzyme engineering.
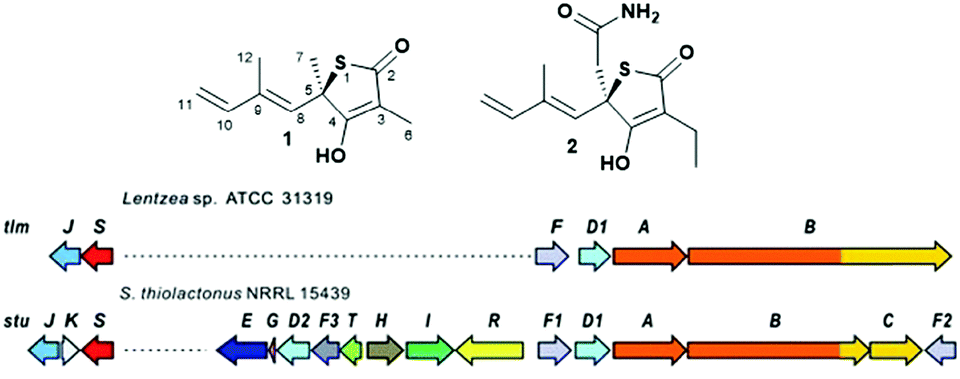 | ||
| Fig. 1 (A) Structure of thiotetronate polyketides thiolactomycin (1) and Tü 3010 (2) and (B) overview of their recently elucidated gene clusters from Lentzea sp. and S. thiolactonus NRRL 15439, respectively.5,6 Legend: J = tRNA-specific thiouridylase; K = N-acetylmuramoyl-L-alanine amidase; S = NifS-like cysteine desulfurase; E = asparagine synthase; G = ferredoxin; D1 and D2 = cytochrome P450s; F3 = 3-oxoacyl-ACP-synthase III (FabH); T = thioesterase; H = crotonyl-CoA reductase; I = 3-hydroxybutyryl-CoA dehydrogenase; R = LuxR family transcriptional regulator; F, F1 and F2 = 3-oxoacyl-ACP-synthases (FabB/F); A = PKS; B = PKS/NRPS; C = NRPS. | ||
Polyketide biosynthesis proceeds via decarboxylative Claisen condensation of malonate units anchored to acyl carrier proteins (ACPs) with acyl moieties bound to ketosynthase (KS) domains (Fig. 3, path A): the resulting polyketide chain is subjected to variable ketoreduction, dehydration and enoyl reduction after chain extension, before being released from PKSs (e.g. via thioester hydrolysis) and/or post-PKS tailoring (e.g. via methyltransferases, glycosyltransferases,…).17,18 A detailed elucidation of the polyketide assembly is crucial for knowledge-based enzyme and cell engineering aiming at novel polyketide production.19–21 Advances in molecular biology, synthetic chemistry and analytical techniques have greatly aided the probing of PKS pathways in vitro and in vivo.5,6,22–25 Nonetheless, several challenges remain concerning the elucidation of timing and mechanisms related to the polyketide assembly, mostly related to the inaccessibility of biosynthetic intermediates covalently linked to PKSs throughout the product assembly.
We have developed a general strategy for probing polyketide biocatalysis based on the use of synthetic ‘chain termination’ probes: these are nonhydrolysable small-molecule mimics of malonate extender units recruited in polyketide formation that competitively interfere in the decarboxylative Claisen condensation to capture and off-load prematurely truncated biosynthetic intermediates (Fig. 3, path B).26–32 These chemical tools have proved successful for the identification and characterisation of intermediate species from modular26–29,32 and iterative30,31 PKSs, in vitro and in vivo, allowing the gathering of otherwise inaccessible information on the timing and the mechanism of single catalytic events,28,30 and also unveiling novel chemoenzymatic opportunities for the generation of unnatural polyketide derivatives.29 In order to shed light on the nature of the biosynthetic intermediates involved in the thiotetronate assembly and their in vivo processing, we have utilised a range of chain termination probes available in our labs. We initially employed the N-(d3) acetyl methyl ester probes 3a–b, which hydrolyse in vivo to the corresponding ‘active’ carboxylate probes 7a–b (Fig. 2), in microbial fermentations of the wild type thiolactomycin producing Lentzea sp. at variable probe concentrations. In organic extracts of cultures we observed diminished production of thiolactomycin with increasing concentrations of 3a–b, strongly suggesting that the probes were interfering in the thiotetronate assembly. However, only a handful of putative captured intermediates were detected from these substrates (Table S2, ESI†). We therefore turned to second-generation probes featuring N-decanoyl chains, which have recently proved to be more efficient tools in capturing polyketide intermediates in vivo due to their enhanced bioavailability and ease of intermediate isolation.32 In addition to substrates 4–5 previously reported,29–32 we also synthesised and utilised the pseudo-methylmalonyl ester 6 (Scheme S1, ESI†), in order to probe the extender unit substrate specificity of the putative iterative PKSs. In parallel to wild-type strains, mutant strains carrying specific deletions in both Lentzea sp. (ΔtlmA,5 ΔtlmD15 and Δcy) and S. thiolactonus NRRL 15439 (ΔstuB and ΔJΔKΔS),5 as well as Streptomyces avermitilis heterologously expressing the Tü 3010 cluster,5 were utilised in control experiments.
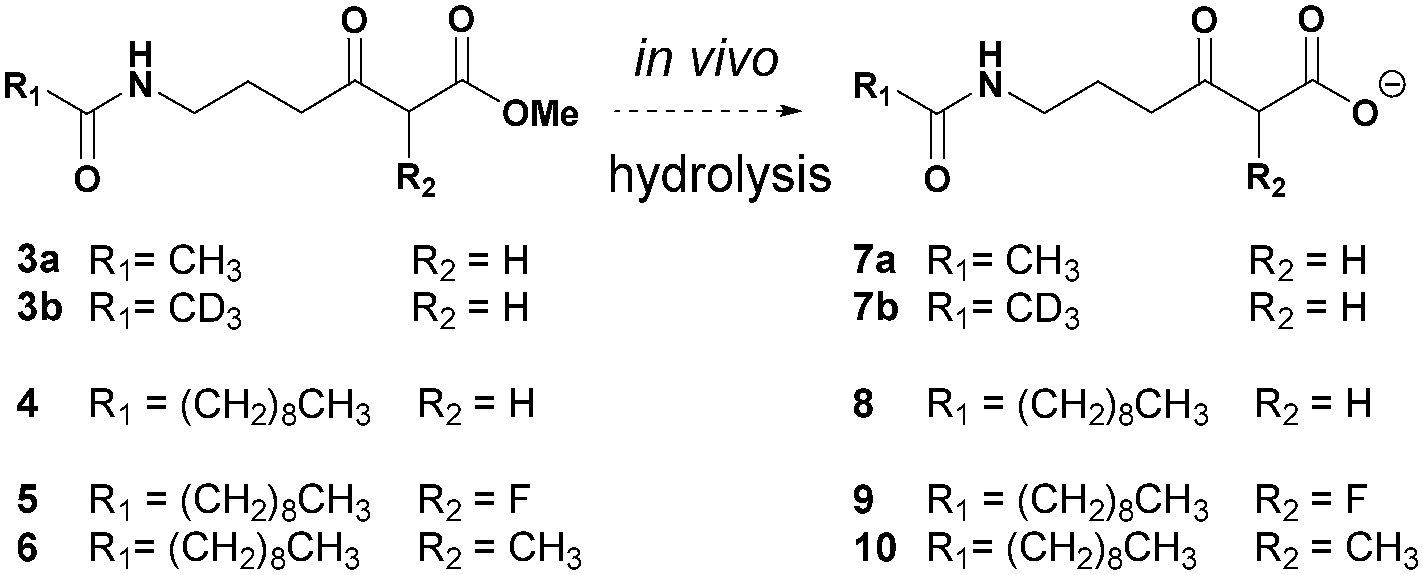 | ||
| Fig. 2 Overview of chemical probes utilised in this study. | ||
An overview of the outcome of feeding experiments of Lentzea sp. and S. thiolactonus strains with compounds 7–10 is given in Fig. 3 and detailed in Tables S2–S5 (ESI†). Although malonate and methylmalonate-mimic probes 8 and 10 preferentially intercepted early stage condensation intermediates, the use of the fluoromalonate-based probe 9 in wild-type Lentzea sp. fermentations captured diketides and triketides presenting various degrees of reduction, as well as putative tetraketides (Table S2, ESI†). These species, directly mirroring the nature of ACP-bound species,5 were characterised by high resolution mass spectrometry tandem experiments, with diagnostic peaks corresponding to the loss of the N-decanoyl moiety and loss of ammonia, and/or cyclic imine formation29–32 (Fig. 3). Although diketide species may be potentially captured from the fatty acid biosynthetic pathway (Table S3, ESI†), the observed triketides and tetraketides were associated exclusively with TLM formation in vivo. In addition to purely polyketide species, a putative S- and O- containing tetraketide species was also captured from Lentzea sp. via9 (Fig. 3C). A parallel investigation of S. thiolactonus wild-type, deletion mutant strains and S. avermitilis heterologously expressing the Tü 3010 cluster5 with methyl ester substrates 4–5 led to the capture of similar putative intermediates, albeit with lower efficiency (Tables S4 and S5, ESI†).
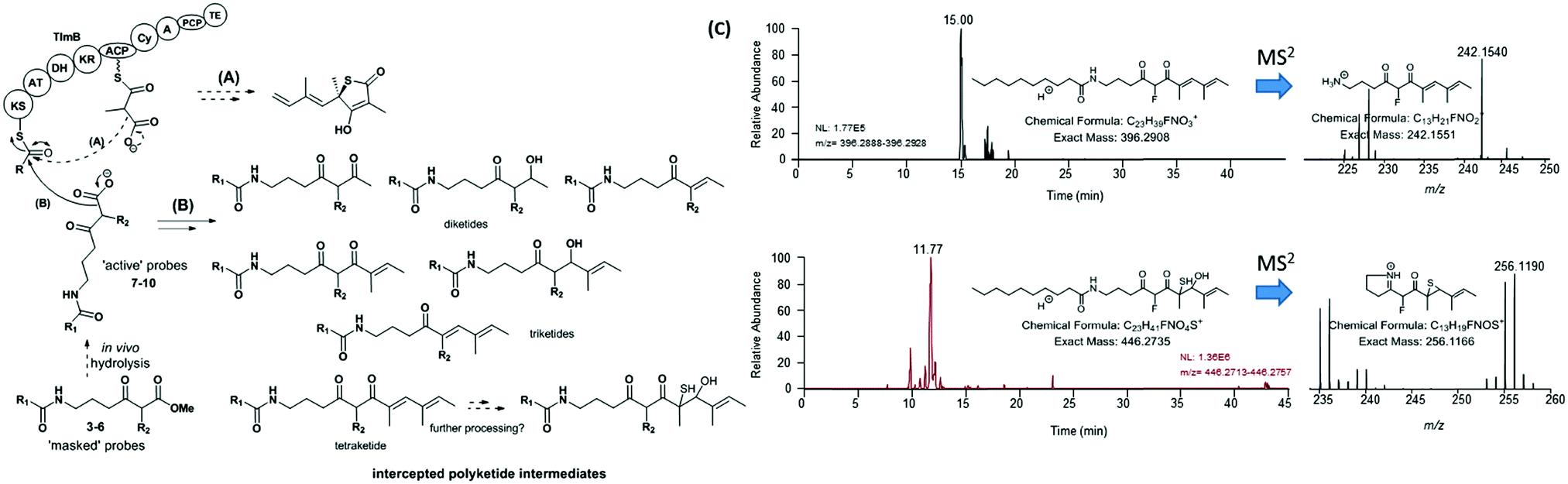 | ||
| Fig. 3 Overview of (A) polyketide assembly and (B) intermediate capture via chemical probes during thiolactomycin (1) assembly in Lentzea sp. (C) Micro LC-HRMS analyses (LTQ-Orbitrap Fusion) of the organic extracts of Lentzea sp. grown in the presence of 5 show the presence of putative tetraketide species captured from the thiolactomycin assembly. Analogous species were captured from cultures of S. thiolactonus strains (Table S4 and Fig. S26, S27, ESI†). The stereochemistry of all the captured species remains undetermined. KS = ketosynthase; AT = acyltransferase; DH = dehydratase; KR = ketoreductase; ACP = acyl carrier protein; Cy = (hetero)cyclisation domain; A = adenylation domain; PCP = peptidyl carrier protein; and TE = thioesterase. | ||
These results taken together constitute the first direct evidence of polyketide chain building and processing involved in thiotetronate bio-assembly. By utilising a variety of chemical probes as pseudo-malonate substrates, preliminary information on substrate specificity and the kinetics of polyketide assembly has been obtained. The pseudo-fluoromalonate 9, generated from in vivo hydrolysis of the corresponding methyl ester 5, proved to be the most efficient tool in capturing almost every intermediate involved in the polyketide TLM assembly. Compared to the malonate and methylmalonate inspired-substrates 4 and 6, we observed enhanced in vivo ester hydrolysis followed by decarboxylation for 5 (Fig. S7, S10 and S20, ESI†): this is possibly due to the electron-withdrawing effect of fluorine at the α-ester carbonyl position, or to the increased probe bioavailability, or to a likely combination of both. On the other hand, 9 did not prove equally efficient in sampling the polyketide chain assembly for Tü 3010, suggesting a much stricter substrate specificity for StuB. By analysing the relative amounts of all the characterised species from Lentzea sp., it appears that, in general, early stage substrate processing is relatively slow, as previously observed for other partially reducing iterative type I synthases (iPKSs),30,31 and that the presence of TlmD1, the TlmB Cy domain and J, K, S proteins is essential for polyketide chain growth and processing beyond the triketide stage. Most type I iPKSs utilise malonate extender units, and methyl groups are introduced into their polyketide products by integral SAM-dependent methyltransferases.33,34 In this respect, TlmB and StuB display unusual substrate specificity in utilising methylmalonate and ethylmalonate extender units, dictated by their respective AT domains. We have shown here that these unusual iPKSs can also process pseudo-malonate, methylmalonate and fluoromalonate substrates at different stages and to various extents. These findings have been corroborated by parallel in vitro investigations with recombinant TlmB in our laboratories.35 We have recently proposed a mechanism for sulphur insertion following the formation of the PKS-bound tetraketide, involving the nonribosomal peptide synthase (NRPS) functionalities encoded in the C-terminal part of TlmB: tetraketide epoxidation (catalysed by TlmD1/StuD1) may precede the formation of a tetraketide–thiirane intermediate from PCP-bound cysteine desulfuration (Tlm/Stu J and S catalysed), followed by heterocyclisation and product release.5 It is tempting to speculate that the putative S- and O- containing tetraketide species characterised from Lentzea sp. and S. thiolactonus wild-type strains may be related to this pathway. Further work will be required to establish the true nature of S-containing intermediates leading to thiotetronate formation. Nevertheless, the deconvolution of the steps of chain assembly reported here has set the stage for the investigation of these post-PKS events.
In summary, we have utilised chemical probes to gather the first information on the intermediates in polyketide assembly for thiotetronate formation in vivo. This represents an important step towards the full elucidation of thiotetronate assembly and the development of synthetic biology-based routes for novel thiotetronate production.
We gratefully acknowledge BBSRC (project grant BB/J007250/1 to M. T.); the Herchel Smith Chair of Biochemistry Fund (award to M. E. Y); EPSRC (DTA PhD studentship to R. J.); the Erasmus programme (exchange bursary to S. H.); the National Science Foundation of China (project grant 31300061 to W. T.); Dr Cleidiane Zampronio (School of Life Sciences, Warwick) for assistance with LC-HRMSn analyses performed on an Orbitrap Fusion instrument; and Dr Lijiang Song for preliminary MS data acquired on a MaXis Bruker Impact instrument.
Notes and references
- E. Larsen, M. R. Wilson and R. E. Taylor, Nat. Prod. Rep., 2015, 32, 1183–1206 RSC.
- F. Hemmerling and F. Hahn, Beilstein J. Org. Chem., 2016, 12, 1512–1550 CrossRef CAS PubMed.
- A. W. Truman, Beilstein J. Org. Chem., 2016, 12, 1250–1268 CrossRef CAS PubMed.
- M. Ma, J. Lohman, T. Liu and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10359–10364 CrossRef CAS PubMed.
- W. Tao, M. E. Yurkovich, S. Wen, K. B. Lebe, M. Samborskyy, Y. Liu, A. Yang, Y. Liu, Y. Ju, Z. Deng, M. Tosin, Y. Sun and P. F. Leadlay, Chem. Sci., 2016, 7, 376–385 RSC.
- X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS PubMed.
- H. Oishi, T. Noto, H. Sasaki, K. Suzuki, T. Hayashi, H. Okazaki, K. Ando and M. Sawada, J. Antibiot., 1982, 35, 391–395 CrossRef CAS PubMed.
- S. Omura, Y. Iwai, A. Nakagawa, R. Iwata, Y. Takahashi, H. Shimizu and H. Tanaka, J. Antibiot., 1983, 36, 109–114 CrossRef CAS PubMed.
- C. Rapp, G. Jung, C. Isselhorst-Scharr and H. Zänhner, Eur. J. Org. Chem., 1988, 1043–1047 CAS.
- T. Sato, K. Suzuki, S. Kadota and M. Iwanami, J. Antibiot., 1989, 42, 890–896 CrossRef CAS PubMed.
- C. A. Machutta, G. R. Bommineni, S. R. Luckner, K. Kapilashrami, B. Ruzsicska, C. Simmerling, C. Kisker and P. J. Tonge, J. Biol. Chem., 2010, 285, 6161–6169 CrossRef CAS PubMed.
- L. Kremer, J. D. Douglas, A. R. Baulard, C. Morehouse, M. R. Guy, D. Alland, L. G. Dover, J. H. Lakey, W. R. Jacobs Jr, P. J. Brennan, D. E. Minnikin and G. S. Besra, J. Biol. Chem., 2000, 275, 16857–16864 CrossRef CAS PubMed.
- S. M. Jones, J. E. Urch, M. Kaiser, R. Brun, J. L. Harwood, C. Berry and I. H. Gilbert, J. Med. Chem., 2005, 48, 5932–5941 CrossRef CAS PubMed.
- G. R. Bommineni, K. Kapilashrami, J. E. Cummings, Y. Lu, S. E. Knudson, C. Gu, S. G. Walker, R. A. Slayden and P. J. Tonge, J. Med. Chem., 2016, 59, 5377–5390 CrossRef CAS PubMed.
- K. Young, H. Jayasuriya, J. G. Ondeyka, K. Herath, C. Zhang, S. Kodali, A. Galgoci, R. Painter, V. Brown-Driver, R. Yamamoto, L. L. Silver, Y. Zheng, J. I. Ventura, J. Sigmund, S. Ha, A. Basilio, F. Vicente, J. R. Tormo, F. Pelaez, P. Youngman, D. Cully, J. F. Barrett, D. Schmatz, S. B. Singh and J. Wang, Antimicrob. Agents Chemother., 2006, 50, 519–526 CrossRef CAS PubMed.
- M. S. Brown, K. Akopiants, D. M. Resceck, H. A. I. McArthur, E. McCormick and K. A. Reynolds, J. Am. Chem. Soc., 2003, 125, 10166–10167 CrossRef CAS PubMed.
- K. J. Weissman, Methods Enzymol., 2009, 459, 3–16 CAS.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4618–4716 CrossRef PubMed.
- C. Hertweck, Trends Biochem. Sci., 2015, 40, 189–240 CrossRef CAS PubMed.
- E. Kim, B. S. Moore and Y. J. Yon, Nat. Chem. Biol., 2015, 11, 649–659 CrossRef CAS PubMed.
- R. Breitling and E. Takano, Curr. Opin. Biotechnol., 2015, 35, 46–51 CrossRef CAS PubMed.
- E. S. Sattely, M. A. Fischbach and C. T. Walsh, Nat. Prod. Rep., 2008, 25, 757–793 RSC.
- S. B. Bumpus and N. L. Kelleher, Curr. Opin. Chem. Biol., 2008, 12, 475–482 CrossRef CAS PubMed.
- E. Esquenazi, Y.-L. Yang, J. Watrous, W. H. Gerwick and P. C. Dorrestein, Nat. Prod. Rep., 2009, 26, 1521–1534 RSC.
- S. Nadmid, A. Plaza, R. Garcia and R. Müller, J. Nat. Prod., 2015, 78, 2023–2028 CrossRef CAS PubMed.
- M. Tosin, L. Betancor, E. Stephens, W. M. A. Li, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2010, 11, 539–546 CrossRef CAS PubMed.
- M. Tosin, Y. Demydchuk, J. S. Parascandolo, C. Blasco-Per, F. J. Leeper and P. F. Leadlay, Chem. Commun., 2011, 47, 3460–3462 RSC.
- M. Tosin, L. Smith and P. F. Leadlay, Angew. Chem., Int. Ed., 2011, 50, 11930–11933 CrossRef CAS PubMed.
- E. Riva, I. Wilkening, S. Gazzola, W. M. A. Li, L. Smith, P. F. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2014, 53, 11944–11949 CrossRef CAS PubMed.
- H. Kage, E. Riva, J. S. Parascandolo, M. F. Kreutzer, M. Tosin and M. Nett, Org. Biomol. Chem., 2015, 13, 11414–11417 CAS.
- J. S. Parascandolo, J. Havemann, H. K. Potter, F. Huang, E. Riva, J. Connolly, I. Wilkening, L. Song, P. F. L. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2016, 55, 3463–3467 CrossRef CAS PubMed.
- I. Wilkening, S. Gazzola, E. Riva, J. S. Parascandolo, L. Song and M. Tosin, Chem. Commun., 2016, 52, 10392–10395 RSC.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 CAS.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
- M. E. Yurkovich, R. Jenkins, Y. Sun, M. Tosin and P. F. Leadlay, Chem. Commun., 2017 10.1039/c6cc09934c.
Footnote |
| † Electronic supplementary information (ESI) available: General methods for the synthesis of chemical probes, Lentzea sp. genetic manipulation and LC-HRMS analysis of the biosynthetic intermediates isolated from Lentzea sp. and S. thiolactonus strains. See DOI: 10.1039/c6cc09933e |
| This journal is © The Royal Society of Chemistry 2017 |