
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn anionic nucleophilic d4 carbyne complex†
Anthony F.
Hill
 * and
Richard Y.
Kong
* and
Richard Y.
Kong
Research School of Chemistry, Australian National University, Canberra, Australian Capital Territory ACT 2601, Australia. E-mail: a.hill@anu.edu.au
First published on 15th December 2016
Abstract
The reaction of the methylidyne complex [W(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH)Br(CO)2(dcpe)] (dcpe = 1,2-bis(dicyclohexylphosphino)ethane) with tBuLi affords the intermediate anionic neopentylidyne complex Li[W(CtBu)(CO)2(dcpe)] which acts as a metal-based nucleophile towards tBuCl, tBuBr, Ph2E2 (E = S, Se, Te) and ClSnMe3 to afford the new carbyne complexes [W(CtBu)(X)(CO)2(dcpe)] (X = Cl, Br, EPh, SnMe3).
CH)Br(CO)2(dcpe)] (dcpe = 1,2-bis(dicyclohexylphosphino)ethane) with tBuLi affords the intermediate anionic neopentylidyne complex Li[W(CtBu)(CO)2(dcpe)] which acts as a metal-based nucleophile towards tBuCl, tBuBr, Ph2E2 (E = S, Se, Te) and ClSnMe3 to afford the new carbyne complexes [W(CtBu)(X)(CO)2(dcpe)] (X = Cl, Br, EPh, SnMe3).
Anionic carbyne complexes remain rare, being limited to Schrock's archetypal [MeN(C2H4)2NMe·Li][Ta(
CtBu)(CH2tBu)3] (1),1 those of the form [M(CR)X4]− (M = Mo, W; X = halide, siloxide, alkoxide)2 in which the coordinatively unsaturated d0-metal3 is electrophilic and Stone's d2-carbaborane complexes [M(CR)(CO)2(L)]− (L = η5-C2B9H9R2′, η6-C2B10R2′ R′ = H, Me)4 in which, whilst much of the negative charge resides in the carbaborane, the metal displays a modest degree of nucleophilicity.
A second rare class of carbyne complexes are those in which the metal has a higher d4 occupancy, employing the [CR]3− formalism,3 which are however limited to late transition metals from groups 8 and 9.5 We report herein the unexpected formation of an anionic d4-early transition metal carbyne complex that serves as a metal-based nucleophile to a wide range of electrophiles.
We have reported extensively on the synthetic utility of the lithiocarbyne complexes [M(CLi)(CO)2(Tp*)] (M = Mo, W; Tp* = hydrotris(dimethylpyrazolyl)borate)6 which are conveniently available via simple lithium/halogen exchange upon treating Lalor's halocarbynes [M(CX)(CO)2(Tp*)]7 with BuLi and which serve as carbon-centred nucleophiles. This allows the construction of a plethora of unusual carbyne complexes that are not available via more conventional protocols. The tungsten derivative had however been previously reported by Templeton to arise from deprotonation of the thermally labile parent methylidyne [W(CH)(CO)2(Tp*)] (estimated pKa = 28.7 in THF)8 and Cummins has separately reported the deprotonation of his methylidyne complex [Mo(CH){NtBuC6H3Me2}3] (estimated pKa = 30 in THF).9 In both cases pKa estimates were not dissimilar to those for notionally isolobal terminal alkynes (e.g., PhCCH: pKa = 28.8 in dmso). Accordingly, with the recent report of the thermally stable methylidyne complex [W(CH)Br(CO)2(dcpe)] (1: dcpe = 1,2-bis(dicyclohexylphosphino)ethane, Scheme 1)10 we naturally considered exploring its deprotonation as a means of generating an anionic carbido complex [W(C)Br(CO)2(dcpe)]−. Indeed the intermediacy of such a species is implicit in the formation of 1 from [W(CSiPh3)Br(CO)2(dcpe)] via fluoride mediated protodesilylation with [nBu4N]F (Scheme 1).
 | ||
| Scheme 1 (i) [nBu4N]F, H2O, –[nBu4N][SiF2Ph3].10 | ||
Treating 1 with a range of bases (nBuLi, PhLi, KH/18-crown-6, LiNiPr2) and ‘super-bases’ (e.g., nBuLi/HOC2H4NMe2) led to disappointing and inconclusive results. Indeed, the only base we could identify that reacted cleanly with 1 was tBuLi and in excess of two equivalents were required. Electrophilic quench of the resulting species with Me3SnCl led to clean formation of a new complex that was not the anticipated stannyl carbyne [W(CSnMe3)Br(CO)2(dcpe)]11 but rather the neopentylidyne complex [W(CtBu)(SnMe3)(CO)2(dcpe)] (2, Scheme 2). Complex 2 was characterised on the basis of structural (see Fig. 1 and ESI†) and spectroscopic (see ESI†) data, the most informative being the triplet (2JPC = 7.5 Hz) carbyne resonance observed at δC = 316.3 that correlates (HMBC) with the tert-butyl resonance observed in the 1H NMR spectrum at δH = 1.30. The single resonance observed at δP = 51.7 in the 31P{1H} NMR spectrum was straddled by satellites due to coupling to 183W (220), 119Sn (151) and 117Sn (145 Hz) nuclei.
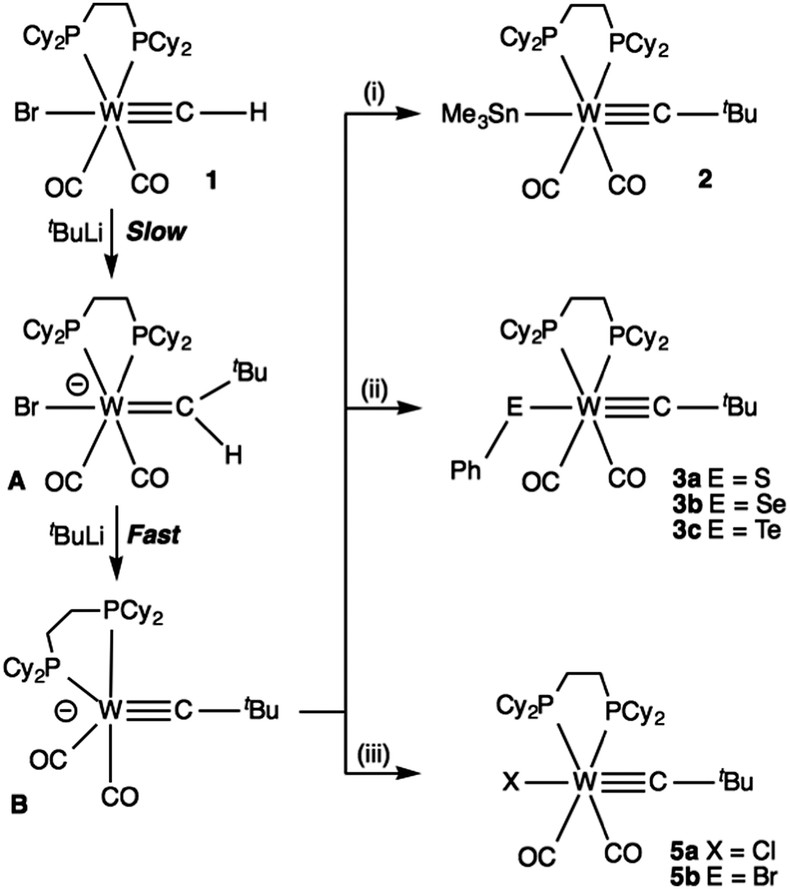 | ||
| Scheme 2 (i) Me3SnCl; (ii) PhEEPh, E = S, Se, Te; (iii) tBuX, X = Cl, Br. All reactions carried out in THF at −78 °C with warming to room temperature after addition of electrophiles (i)–(iii). | ||
 | ||
| Fig. 1 Molecular structure of 2 in a crystal (60% displacement ellipsoids, H-atoms omitted, cyclohexyl groups simplified). Selected bond lengths (Å) and angles: W1–Sn1 2.9332(3), W1–P2 2.5497(9), W1–P3 2.5363(8), W1–C1 1.834(4), Sn1–W1–C1 160.73(12), W1–C1–C2 164.3(3). | ||
The molecular structure of 2 (Fig. 1) is based on a pseudo-octahedral tungsten with distortion from ideal geometry being attributable to inter-ligand repulsions. The WC bond-length of W1–C1 1.834(4) Å, is unremarkable, falling within the normal range for neopentylidyne complexes although all previous structural studies involved this ligand bound to high-valent tungsten. While there are copious structural data available for tungsten stannyl complexes, the only two trimethylstannyl examples [W(SnMe3)(CO)2(η-C5H4SnMe3)] (W–Sn = 2.8481(5) Å)12 and [W(SnMe3)(CO)5]− (2.810(8) Å)13 have far shorter W–Sn bonds than found in 2 (2.9332(3) Å), a manifestation of the superlative trans influence of carbyne ligands.
The reaction proved to be comparatively general for a range of electrophiles (Scheme 2). Phenylchalcogenolato complexes [W(CtBu)(EPh)(CO)2(dcpe)] (E = S 3a, Se 3b, Te 3c; see ESI†) were obtained by employing PhEEPh as the electrophilic quench and were each structurally characterised. Structural data for 3b (Fig. 2, for depictions of 3a and 3c see ESI†) typify those for the series, the notable features being an insignificant impact on the WC bond length, from 1.821(5) Å for 3a to 1.823(4) Å for 3c. In a similar manner, neither the νCO frequencies nor the chemical shift of the carbyne carbon were particularly sensitive to variations in the chalcogen (Table 1). The W–E–C angle decreases only marginally from 107.4 for 3a to 104.8(1)° for 3c and while this modest change might appear consistent with the usual trend for reduced s-character with heavier chalcogens, it should be pointed out that both 3b and isomorphous 3c participate in incipient intermolecular π-stacking of the phenyl rings (absent for 3a, see ESI†) so as to cloud any such interpretation.
 | ||
| Fig. 2 Molecular structure of 3b in a crystal (60% displacement ellipsoids, H-atoms omitted, cyclohexyl groups simplified). Selected bond lengths (Å) and angles: W1–Se2 2.7467(7), W1–P3 2.5489(14), W1–P4 2.5696(15), W1–C6 1.826(7), Se2–C13 1.927(6), C6–W1–Se2 167.9(2), C7–C6–W1 167.4(5), C13–Se2–W1 107.5(2). | ||
CtBu)X(CO)2(dcpe)]
| X | ν CO | k CO |
δ
C (WC) |
2 J PC |
|---|---|---|---|---|
| [cm−1] | [Ncm−1] | [ppm] | [Hz] | |
| a Cotton–Kraihanzel force constant for cis-dicarbonyl. b Measured in toluene. c Measured in C6H6. d Measured in CH2Cl2. e Measured in CDCl3. | ||||
| SnMe32 | 1961, 1899b | 15.02 | 317.2c | 7.5 |
| SPh 3a | 1997, 1928d | 15.53 | 297.3e | 9.2 |
| SePh 3b | 1984, 1915b | 15.33 | 298.8e | 8.7 |
| TePh 3c | 1981, 1914b | 15.30 | 301.4e | 8.4 |
| Cl 5a | 1991, 1915d | 15.39 | 289.5e | 7.6 |
| Br 5b | 1978, 1908d | 15.23 | 290.4e | 7.5 |
Treating the intermediate with tBuBr or tBuCl as mild sources of HX (X = Cl, Br) in E2 reactions results not in the tertiary alkyl or hydrido [W(CtBu)H(CO)2(dcpe)] (4) derivatives, but rather the halides [W(CtBu)X(CO)2(dcpe)] (X = Cl 5a, Br 5b), both of which were structurally characterised (see ESI†). Berke has described a range of hydrido-carbyne complexes of the form [W(CtBu)H(L)4] (L4 = various combinations of CO, P(OMe)3, PMe3)14 and shown, e.g., that phenol is a strong enough acid (pKa 10) to cleave the hydride ligands so we may assume that 4 is viable but presumably is rapidly converted to the halide complexes by excess alkyl halide.
As noted above for 2, the yield is maximised when two or more equivalents of tBuLi are employed and with only one equivalent we have been unable to isolate or spectroscopically identify any intermediates. We contend therefore that a two step process operates that involves slow initial nucleophilic attack at the methylidyne carbon of 1 by tBuLi to afford the anionic neopentylidene Li[W(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHtBu)Br(CO)2(dcpe)] (A, Scheme 2) which then undergoes a rapid dehydrohalogenation by a second equivalent of tBuLi to afford the anionic neopentylidyne complex Li[W(CtBu)(CO)2(dcpe)] (B), precluding the accumulation of A. We have been unable to isolate ‘B’ which decomposes below room temperature under the conditions of its formation. Indeed the reactions involving tBuX (X = Cl, Br) were specifically motivated by attempts to quench unreacted excess tBuLi.
CHtBu)Br(CO)2(dcpe)] (A, Scheme 2) which then undergoes a rapid dehydrohalogenation by a second equivalent of tBuLi to afford the anionic neopentylidyne complex Li[W(CtBu)(CO)2(dcpe)] (B), precluding the accumulation of A. We have been unable to isolate ‘B’ which decomposes below room temperature under the conditions of its formation. Indeed the reactions involving tBuX (X = Cl, Br) were specifically motivated by attempts to quench unreacted excess tBuLi.
The intermediate in the absence of an electrophilic trapping agent decomposes extremely rapidly near room temperature and it has not been possible to acquire useful infrared data, however it is unlikely that this technique would not reliably discriminate between the two alternatives. In situ NMR spectroscopy (d8-THF) proved somewhat more informative. When 1 was treated with less than 2 equivalents of tBuLi a solution of the intermediate could be maintained at −62 °C long enough to acquire both 31P, 1H and 1H–13C HMBC spectra. The 31P{1H} NMR spectrum (Fig. 3) reveals in addition to unreacted 1, a second predominant compound at δP = 90.2 ppm (1JWP = 215 Hz), with no evidence for any other major compounds being present.
 | ||
| Fig. 3 31P{1H} NMR spectrum (in situ, d8-THF) of the reaction of 1 (blue) with ca. 1.5 equivalents of tBuLi at −30 °C to afford intermediate B (pink). | ||
No resonances were observed in the alkylidene region of the 1H NMR spectrum (δH = 10–25 ppm). The 1H–13C HMBC spectrum (see ESI†) revealed a resonance at δC = 322 in the alkylidyne region which correlates with that observed for the tBu group (CH3) at δH = 1.06 but to no other (i.e., alkylidene CH type, δH > 10) resonance. These data together all support the accumulation of alkylidyne intermediate B, which is formed rapidly from alkylidene A which does not accumulate in spectroscopically determinable amounts.
Allowing the sample to warm to room to room temperature results in rapid formation of free dcpe (δP = 0.92, no coupling to 183W) which is perhaps to be expected since trialkylphosphines do not generally bind strongly to anionic metal centres. The rapid deprotonation is perhaps surprising for an anionic neopentylidene in a low oxidation state, but has precedent for high oxidation state systems, e.g., [W(CHPh)Cl2(CO)(PMe3)2],15 which is dehydrohalogenated by pyrollidinyl cyclopentene, though in that case an agostic C–H–W interaction contributes to the acidity.16 For the deprotonation of B it is therefore likely that initial halide dissociation affords the neutral α-agostic neopentylidene [W(CHtBu)(CO)2(dcpe)] or its carbyne tautomer [W(CtBu)H(CO)2(dcpe)], either of which would have enhanced acidity. The sterically modest methylidyne would appear to allow nucleophilic attack (rather than deprotonation) while, e.g., the more congested benzylidene [W(CPh)Br(CO)2(dcpe)] (6) (obtained from [W(CPh)Br(CO)2(picoline)2] and dcpe), fails to undergo a similar reaction. The HOMO of the simpler model complex [W(CMe)(CO)2(dmpe)]− (see ESI†) was found to be primarily tungsten-based, and to protrude from a trigonal bipyramidal tungsten opposite to the equatorial coordinated ethylidyne ligand, thereby accounting for the observed stereochemistry of products (2–5; electrophile trans to CtBu) from electrophilic attack at B. While we are confident in the identity of B, we are unable to discount an alternative route that might also lead to it. Lithium–halogen exchange reactions between tBuLi and aryl bromides are generally considered to proceed via rapid single electron transfer events. Similar processes with 1 involving reduction, hydrogen abstraction and capture of the comparatively long-lived tert-butyl radical may well operate and account for the failure to observe A.
To conclude, whilst the terminal methylidyne 1 is against all expectations not susceptible to deprotonation, it appears readily converted to the first example of an early transition metal high d-occupancy carbyne complex, which displays synthetic versatility through metal-based nucleophilicity. Not all electrophiles met with success in our hands. Thus as noted, treating B with tBuX affords halo-neopentylidyne complexes [W(CtBu)X(CO)2(dcpe)] rather than the desired alkyl or hydrido complexes and NH4Cl also failed to provide an isolable hydrido derivative. The reaction of B with Ph3SnCl provided primarily the chloro complex 5a, presumably via single electron transfer processes that are favoured by the steric incompatibility of both reagents. We were similarly unable to isolate methyl, benzyl or trifluoromethyl complexes from reactions of B with CH3I, PhCH2Cl or (CF3CO)2O though we do not rule out their initial formation. Nevertheless, the species B does afford access to new carbyne complexes with bonds between tungsten and halogens, tin, sulphur, selenium and tellurium.
We gratefully acknowledge the financial support of the Australian Research Council (DP130102598).
Notes and references
- L. J. Guggenberger and R. R. Schrock, J. Am. Chem. Soc., 1975, 97, 2935 CrossRef CAS.
- (a) J. H. Wengrovius, J. Sancho and R. R. Schrock, J. Am. Chem. Soc., 1981, 103, 3932–3934 CrossRef CAS; (b) L. Giannini, E. Solari, S. Dovesi, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1999, 121, 2784–2796 CrossRef CAS; (c) J. Heppekausen, R. Stade, R. Goddard and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 11045–11057 CrossRef CAS PubMed; (d) A. D. Lackner and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 12814–12818 CrossRef CAS PubMed; (e) C. K. Simpson, R. E. Da Re, T. P. Pollagi, I. M. Steele, R. F. Dallinger and M. D. Hopkins, Inorg. Chim. Acta, 2003, 345, 309–319 CrossRef CAS; (f) M. E. O’Reilly, S. S. Nadif, I. Ghiviriga, K. A. Abboud and A. S. Veige, Organometallics, 2014, 33, 836–839 CrossRef; (g) S. Venkatramani, N. B. Huff, M. T. Jan, I. Ghiviriga, K. A. Abboud and A. S. Veige, Organometallics, 2015, 34, 2841–2848 CrossRef CAS.
- Herein we ascribe a trianionic charge to the carbyne ligand to arrive at oxidation state and the d-configurations derived from them. Other schools ascribe a cationic charge to the carbyne ligand (CF+ being isoelectronic with NO+). The choice is arbitrary when discussing highly covalent MC multiple bonds so long as a consistent notation is adhered to.
- (a) S. A. Brew and F. G. A. Stone, Adv. Organomet. Chem., 1993, 35, 135–186 CrossRef CAS; (b) F. G. A. Stone, Adv. Organomet. Chem., 1990, 31, 53–89 CrossRef CAS.
- (a) G. R. Clark, K. Marsden, W. R. Roper and L. J. Wright, J. Am. Chem. Soc., 1980, 102, 6570–6571 CrossRef CAS; (b) E. O. Fischer, J. Schneider and D. Neugebauer, Angew. Chem., Int. Ed. Engl., 1984, 23, 820–821 CrossRef; (c) D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12580–12583 CrossRef CAS PubMed; (d) Y. Lee and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 4438–4446 CrossRef CAS PubMed; (e) K. Ilg and H. Werner, Chem. – Eur. J., 2001, 7, 4633–4639 CrossRef CAS PubMed; (f) T. Rappert, O. Nürnberg, N. Mahr, J. Wolf and H. Werner, Organometallics, 1992, 11, 4156–4164 CrossRef CAS; (g) S. Anderson and A. F. Hill, Organometallics, 1995, 14, 1562–1564 CrossRef CAS; (h) R. B. Bedford, A. F. Hill, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1996, 35, 95–97 CrossRef CAS; (i) H. F. Luecke and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 11008–11009 CrossRef CAS; (j) S. R. Caskey, M. H. Stewart, Y. J. Ahn, M. J. A. Johnson and J. W. Kampf, Organometallics, 2005, 24, 6074–6076 CrossRef CAS; (k) J. C. Conrad, D. Amoroso, P. Czechura, G. P. A. Yap and D. E. Fogg, Organometallics, 2003, 22, 3634–3636 CrossRef CAS; (l) J. N. Coalter III, J. D. Bollinger, O. Eisenstein and K. G. Caulton, New J. Chem., 2000, 24, 925–927 RSC.
- (a) A. L. Colebatch and A. F. Hill, Organometallics, 2016, 35, 2249–2255 CrossRef CAS; (b) A. L. Colebatch, A. F. Hill and M. Sharma, Organometallics, 2015, 34, 2165–2182 CrossRef CAS; (c) A. L. Colebatch and A. F. Hill, J. Am. Chem. Soc., 2014, 136, 17442–17445 CrossRef CAS PubMed; (d) A. F. Hill and R. Shang, Organometallics, 2012, 31, 4635–4638 CrossRef CAS; (e) A. F. Hill, R. Shang and A. C. Willis, Organometallics, 2011, 30, 3237–3241 CrossRef CAS; (f) R. L. Cordiner, A. F. Hill, R. Shang and A. C. Willis, Organometallics, 2011, 30, 139–144 CrossRef CAS; (g) A. L. Colebatch, A. F. Hill, R. Shang and A. C. Willis, Organometallics, 2010, 29, 6482–6487 CrossRef CAS; (h) R. L. Cordiner, A. F. Hill and J. Wagler, Organometallics, 2008, 27, 5177–5179 CrossRef CAS.
- (a) T. Desmond, F. J. Lalor, G. Ferbuson and M. Parvez, J. Chem. Soc., Chem. Commun., 1983, 457–459 RSC; (b) F. J. Lalor, T. J. Desmond, G. M. Cotter, C. A. Shanahan, G. Ferguson, M. Parvez and B. Ruhl, J. Chem. Soc., Dalton Trans., 1995, 1709–1726 RSC.
- A. E. Enriquez, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 2001, 123, 4992–5002 CrossRef CAS PubMed.
- J. B. Greco, J. C. Peters, T. A. Baker, W. M. Davis, C. C. Cummins and G. Wu, J. Am. Chem. Soc., 2001, 123, 5003–5013 CrossRef CAS PubMed.
- A. F. Hill, J. S. Ward and Y. Xiong, Organometallics, 2015, 34, 5057–5064 CrossRef CAS.
- The anionic carbido complex Li[W(C)(CO)2(Tp*)] reacts with Me3SnCl to afford a stannylcarbyne complex [W(CSnMe3)(CO)2(Tp*)]: E. S. Borren, A. F. Hill, R. Shang, M. Sharma and A. C. Willis, J. Am. Chem. Soc., 2013, 135, 4942–4945 CrossRef CAS PubMed.
- H. Bera, H. Braunschweig, R. Dorfler, T. Kupfer, K. Radacki and F. Seeler, Organometallics, 2010, 29, 5111–5120 CrossRef CAS.
- D. J. Darensbourg, C. G. Bauch, J. H. Reibenspies and A. L. Rheingold, Inorg. Chem., 1988, 27, 4203–4207 CrossRef.
- E. Bannwart, H. Jacobsen, F. Furno and H. Berke, Organometallics, 2000, 19, 3605–3619 CrossRef CAS.
- (a) C. M. Bastos, K. S. Lee, M. A. Kjelsberg and A. Mayr, Inorg. Chim. Acta, 1998, 279, 7–23 CrossRef CAS; (b) A. Mayr, M. F. Asaro, M. A. Kjelsberg, K. S. Lee and D. Van Engen, Organometallics, 1987, 6, 432–434 CrossRef CAS.
- Such an α-agostic C–H–W interaction may be discounted in the case of B due to the tungsten being coordinatively saturated.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic and crystallographic data. CCDC 1515542 and 1515557–1515561. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc09718a |
| This journal is © The Royal Society of Chemistry 2017 |