
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced photocatalytic activity of gold nanoparticles driven by supramolecular host–guest chemistry†
Marc
Padilla
,
Francesca
Peccati
,
José Luis
Bourdelande
,
Xavier
Solans-Monfort
,
Gonzalo
Guirado
,
Mariona
Sodupe
* and
Jordi
Hernando
 *
*
Departament de Química, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Spain. E-mail: Mariona.Sodupe@uab.cat; Jordi.Hernando@uab.cat
First published on 23rd January 2017
Abstract
Functionalization of gold nanoparticles with supramolecular hosts allows their plasmon-based photocatalytic activity to be enhanced. This is mainly ascribed to the formation of labile host–guest complexes with the reagent molecules on the metal surface, thus promoting nanoparticle-substrate approximation without interfering with the light-induced catalytic process.
Noble metal nanoparticles (NMNPs) exhibit unique optical properties arising from the excitation of their localized surface plasmon resonance,1 and are widely exploited in a plethora of applications.2 Among them, the use of NMNPs as photocatalysts is currently gaining increasing interest,3 and to date, their photocatalytic activity has been demonstrated for a number of chemical reactions, such as reduction,4 oxidation5 and dissociation6 processes.
Three mechanisms account for the photocatalytic activity of plasmonic NMNPs:3 (a) near field enhancement, which leads to a gigantic increase of the excitation light intensity around the particles and, therefore, of the linear and nonlinear absorption efficiencies of nearby photoreagents; (b) the photothermal effect, i.e. the conversion of light into heat at their surface, which can promote thermally-induced chemical processes; (c) the generation of hot charge carriers, which can be injected into surrounding molecules to trigger redox reactions. Despite their different natures, all of these mechanisms share a common feature: their effects are highly local and, as a result, they are only operative at nanometer-scale distances from the NPs.3 Consequently, a maximal approach of the substrate molecules is required to attain optimal photocatalytic efficiencies.
Normally, this is achieved by diffusion of reactants from the bulk solution to the surface of the photocatalyst, which typically consists of noncoated nanoparticles directly dispersed in the reaction medium or immobilized onto solid supports.4–6 In contrast to what is often observed for freely standing, naked particles, the latter enables their stabilization and reuse, though at the expense of decreasing the catalytic area.4–6 On the other hand, much higher catalytic efficiency could be achieved if reactants were initially tethered to the NMNP surface, thus warranting minimal separation and large plasmonic effects.6b However, this would limit the concentration of substrate molecules that could be photoreacted. To overcome these limitations, herein we propose a novel scheme for plasmon-based photocatalysis relying on supramolecular host–guest chemistry. In our approach, NMNPs are coated with macrocyclic ligands that form supramolecular complexes with the reagent molecules, thereby favoring approximation to their surface and, as a result, leading to larger plasmon-based photocatalytic activities with respect to nonfunctionalized particles (Fig. 1).
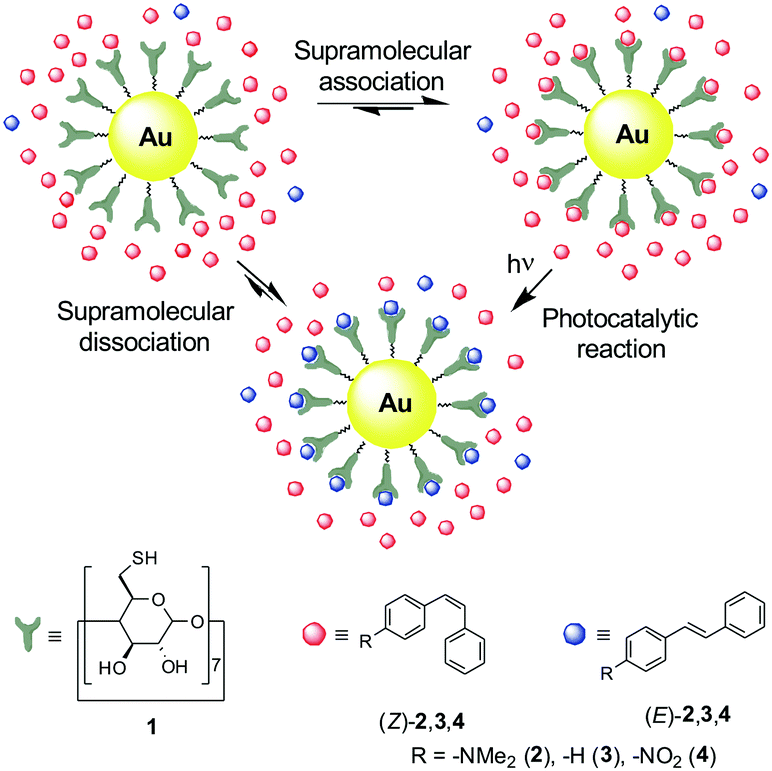 | ||
| Fig. 1 Strategy devised to supramolecularly enhance the photocatalytic activity of gold nanoparticles. | ||
To validate our methodology, we devised a photocatalytic system composed of nonsupported gold nanoparticles coated with β-cyclodextrin (β-CD) derivative 1 (AuNP@1, Fig. 1). While colloidal AuNPs are well-known plasmonic nanostructures with visible-light activity,1 β-CD is an optically inactive macrocyclic host that forms 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complexes in aqueous media with a variety of organic molecules.7 These complexes are mainly driven by hydrophobic effects and present low-to-intermediate association constants (typically, Ka < 104 M−1).7 Therefore, labile host–guest systems could be formed between AuNP@1 and many potential reactants (and products) in water, thus enhancing plasmon-based photocatalysis while ensuring continuous renewal of the complexed molecular substrates to maximize the overall reaction yield. In addition, binding of heptathiolated β-CD 1 to the nanoparticle surface via multiple sulfur bonds must provide AuNP@1 with high photochemical stability, which should allow for long irradiation times and photocatalytic cycles without requiring the use of supports.
:1 inclusion complexes in aqueous media with a variety of organic molecules.7 These complexes are mainly driven by hydrophobic effects and present low-to-intermediate association constants (typically, Ka < 104 M−1).7 Therefore, labile host–guest systems could be formed between AuNP@1 and many potential reactants (and products) in water, thus enhancing plasmon-based photocatalysis while ensuring continuous renewal of the complexed molecular substrates to maximize the overall reaction yield. In addition, binding of heptathiolated β-CD 1 to the nanoparticle surface via multiple sulfur bonds must provide AuNP@1 with high photochemical stability, which should allow for long irradiation times and photocatalytic cycles without requiring the use of supports.
AuNP@1 was prepared through a ligand exchange process between citrate-capped AuNPs and previously synthesized 1 (see the ESI†). 1H NMR and 1D DOSY 1H NMR spectra confirmed surface functionalization with the target β-CD derivative and removal of the excess unbound 1 molecules (Fig. S1 in the ESI†). The resulting nanoparticles preserved the shape, size (diameter = 14 ± 2 nm, Fig. S2 in the ESI†) and extinction spectrum (λmax = 523 nm in water, Fig. S4 in ESI†) of the precursor AuNPs. In addition, replacement of citrate by 1 notably increased their stability in both aqueous and organic media (Fig. S4 in ESI†).
As a proof of concept, we chose a unimolecular reaction to test AuNP@1 photocatalysis: the Z → E isomerization of stilbenes, a relevant family of photoswitches8 capable of supramolecularly interacting with CDs.9 This is the case of 4-dimethylaminostilbene (2), stilbene (3) and 4-nitrostilbene (4), three stilbene derivatives with differing electronic properties, both the stereoisomers of which form 1:1 complexes in aqueous media with β-CD and moderate association constants (Fig. 1 and Table S1 in the ESI†). As for most stilbenes,8 the photoswitching performance of 2–4 presents several downsides, two of which are the need for highly energetic UV light to induce Z → E isomerization (254 nm) and the moderate interconversion yields obtained (<65%, Table S2 in the ESI†). Alternatively, stilbene Z → E isomerization can take place thermally10 or upon charge transfer,11 which could enable plasmon-based photocatalysis of this process via photothermal effects and/or hot charge carrier injection using AuNPs and visible light. Actually, (Z)-2 isomerized upon bulk heating, a behavior that was not suppressed by supramolecular effects upon addition of β-CD (Table S3 in the ESI†). This, in combination with its ∼10-fold larger association constant with β-CD, prompted us to select this compound as the benchmark system to validate our supramolecular approach to plasmon-based photocatalysis.
Table 1 displays the results obtained for the photocatalytic studies of (Z)-2 isomerization. They were performed at room temperature and under pulsed laser light at 532 nm, which is absorbed by none of the isomers of 2 (Fig. S6 in the ESI†). Control experiments showed insignificant Z → E conversions in the absence of catalyst and/or illumination, thus proving thermal and photochemical stability of (Z)-2 under our photocatalytic conditions (entries 1–3). Importantly, this is in contrast to the behavior of (Z)-azobenzenes, whose isomerization is catalyzed by AuNPs even in the dark.12 In the case of 2, both AuNP@1 addition and green light illumination were required to observe Z → E conversion (Fig. S8 in ESI†), while the formation of typical by-products of direct photo-isomerization of stilbene (e.g. phenantrenes8) was not detected. The Z → E yield was found to depend on catalyst concentration, laser power and irradiation time (entries 4–10), and a maximum 56% conversion to (E)-2 was found at cAuNP@1 = 45 nM, 300 mW power and 2 h of irradiation. This notably enhanced the results obtained upon direct excitation of (Z)-2 with UV light (41%). Longer irradiation times did not improve the yield, while fast AuNP@1 degradation was observed at higher powers (600 mW).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | c catalyst (nM) | Power (mW) | Time (h) | Yieldc (%) |
| a c (Z)- 2 = 11.3 mM, 6% acetonitrile in water, Ar atmosphere and room temperature. b Irradiation with a 6 ns pulsed laser at 10 Hz repetition rate. c Determined from 1H NMR after treatment of the reaction mixture (see ESI). d Average value for 3 independent experiments (±5%). e Upon AuNP@1 recycling, isomerization yields of 58 and 61% were determined for two additional photocatalytic cycles. f c 1-AdNH2 = 80 mM. g c β-CD = 100 mM. | |||||
| 1 | — | — | — | 2 | 4 |
| 2 | — | — | 300 | 2 | 2 |
| 3 | AuNP@1 | 45 | — | 2 | 6 |
| 4 | AuNP@1 | 18 | 300 | 0.5 | 5 |
| 5 | AuNP@1 | 45 | 150 | 0.5 | 5 |
| 6 | AuNP@1 | 45 | 150 | 1 | 11 |
| 7 | AuNP@1 | 45 | 150 | 2 | 16 |
| 8 | AuNP@1 | 45 | 300 | 0.5 | 18 |
| 9 | AuNP@1 | 45 | 300 | 1 | 37 |
| 10 | AuNP@1 | 45 | 300 | 2 | 56d,e |
| 11 | AuNP@1 + 1-AdNH2f | 45 | 300 | 2 | 17 |
| 12 | AuNP@1 + β-CDg | 45 | 300 | 2 | 16 |
| 13 | AuNP@citrate | 45 | 300 | 2 | 15 |
| 14 | AuNP@PVP | 45 | 300 | 2 | 3 |
Noticeably, catalyst reuse was enabled thanks to the enhanced NP stability provided by the β-CD ligands. However, two factors ultimately limited the recyclability of our catalytic system. First, AuNP@1 recovery from the reaction mixture by centrifugation was not fully efficient and a loss of ∼30% of the catalyst mass was observed. In spite of this, when correcting for the amount of catalyst recovered, nearly no variation in conversion yield was observed after 3 consecutive catalytic cycles (entry 10). In addition, slow photodegradation of the AuNPs into smaller nanostructures (∼2–7 nm in size) was also found after prolonged irradiation at 300 mW, a process that affected about 1–6% of the initial catalyst mass (Fig. S9 in ESI†). To ensure that it did not alter our measurements, ∼5 nm-in-diameter AuNPs coated with 1 were prepared separately (AuNP-s@1, Fig. S5 in the ESI†) and their photocatalytic efficiencies were assessed independently. Under the best conditions found for the larger particles, the use of AuNP-s@1 led to significantly lower conversion yields (<20%, see Table S5 in the ESI†). This unambiguously proves that the Z → E isomerization efficiencies determined for AuNP@1 arise from the photocatalytic activity of the initial ∼15 nm-in-diameter particles prepared.
Once the plasmon-based photocatalytic activity of AuNP@1 on (Z)-2 isomerization was proven, we investigated the supra-molecular effects imparted by the β-CD ligand. For this we first executed additional experiments with AuNP@1 where catalyst–substrate recognition was hindered by adding a large excess of: (a) 1-adamantylamine (1-AdNH2), which forms highly stable complexes with β-CD (Ka > 103 M−1) (entry 11);13 (b) free non-thiolated β-CD (entry 12). In both cases, ∼3.5-fold lower Z → E conversions were found, thus supporting the occurrence of supramolecular effects on our catalytic system. Moreover, other experiments were conducted using CD-free AuNPs (∼15 nm in diameter) that do not supramolecularly interact with 2 or even preclude direct catalyst–substrate contact: (c) AuNP@citrate (entry 13); (d) AuNPs coated with a very thin shell (1.4 nm) of polyvinylpyrrolidone (AuNP@PVP, see Fig. S2–S4 in the ESI†, entry 14). Low-to-negligible stilbene isomerization was observed in these cases, which further proves enhancement of AuNP@1 photocatalysis by supramolecular chemistry. In addition, AuNP@citrate and AuNP@PVP could not be recycled due to their poorer photostability under our experimental conditions, thereby demonstrating the added value of using heptathiolated ligand 1 as a photocatalyst stabilizer.
To gain a deeper insight into the supramolecularly-enhanced activity of AuNP@1, we expanded our studies to stilbenes 3 and 4. For these compounds, remarkably lower photocatalytic yields were measured (<15%), which however were ca. 2.5-fold larger than those registered when using AuNP@citrate (Fig. 2A and Table S4 in the ESI†). This proves supramolecular effects even on the photocatalytic isomerization of (Z)-3 and (Z)-4 despite their lower association constants with β-CD (Table S1 in the ESI†), a result ascribed to the similar amounts of reactive CD–stilbene complexes expected to form in all the cases regardless of the distinct Ka values (∼0.1% in the steady state under our conditions). Actually, the dramatic changes in photocatalytic activity observed for 2–4 cannot be explained on the sole basis of their different supramolecular association constants, since clearly larger photocatalytic efficiencies were also found for (Z)-2 with AuNP@citrate. Therefore, a crucial role must also be played by the intrinsic capacity of AuNPs to photoinduce the isomerization of electronically-different stilbenes, which must be ultimately related with the underlying catalytic mechanism.
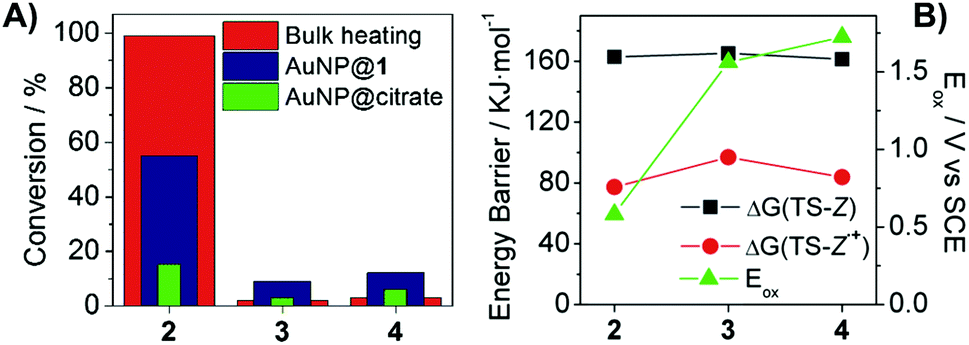 | ||
| Fig. 2 (A) Z → E conversions for 2–4 upon bulk heating without AuNPs (423 K, 30 min, DMSO) and photocatalysis with AuNP@1 and AuNP@citrate (300 mW, 2 h). (B) Theoretical isomerization energy barriers computed for the neutral (ΔG(TS-Z)) and radical cation forms (ΔG(TS-Z˙+)) of (Z)-2–4, and experimental oxidation potentials (Eox) of these compounds. | ||
AuNP@1 photocatalysis of stilbene isomerization could be attributed to a photothermal mechanism. This was explored by bulk heating (Z)-2–4 in the dark under different conditions (Table S3 in the ESI†). In the absence of AuNPs, thermal Z → E conversion was only observed for 2, a behavior that qualitatively resembles our photocatalytic results (Fig. 2A). Nevertheless, the kinetics of the thermal isomerization of (Z)-2 does not correspond to that of a simple unimolecular reaction, but to a more complex mechanism (Fig. S7 in the ESI†), which prevents ascribing plasmon-based stilbene Z → E conversion to pure photothermal effects. On the other hand, thermal isomerization was measured for both (Z)-2 and (Z)-4 after AuNP@1 addition, in contrast to our photocatalytic data. These results, however, are affected by the rapid degradation found for AuNP@1 upon bulk heating, which might produce a highly active gold catalytic species accounting for stilbene isomerization in the dark. To better assess the photothermal effects on AuNP@1 photocatalysis, previously calibrated broken symmetry density functional theory calculations were performed to compute the energy barrier for the thermal Z → E isomerization of 2–4 (see the ESI† for details). Large and very similar barriers were obtained for the three compounds (Fig. 2B). Therefore, no significant differences in Z → E conversion should be expected for 2–4 if the AuNP@1 photocatalytic effect was purely photothermal, which is in striking contrast with our experimental data.
An alternative photocatalytic mechanism would be near-field enhancement of the two-photon (2P) isomerization of (Z)-stilbenes with visible light, as reported for azobenzenes.14a However, (E)-stilbenes present larger 2P absorption cross-sections than their (Z)-isomers, which makes E → Z isomerization the most favored nonlinear optical process under irradiation at 532 nm.14b Since no E → Z conversion was observed for (E)-2 when subjected to the best photocatalytic conditions found for (Z)-2 (Table S4 in the ESI†), we concluded that the AuNP@1-induced Z → E stilbene conversion cannot be ascribed to near-field enhancement of 2P isomerization.
Finally, we explored a third type of mechanism for AuNP@1 photocatalysis, which involves hot charge carrier injection from the particles followed by isomerization of the resulting stilbene ion. In particular, our attention focused on analyzing the effect of hole injection from AuNP@1 to the substrate, since the largest yields were measured for electron-rich (Z)-2. Two main parameters were evaluated to assess the viability of this mechanism: (a) the oxidation tendency of 2–4, which was determined both by cyclic voltammetry and computationally (Fig. 2B and Tables S7, S8 and Fig. S10 in the ESI†); (b) the energy barrier for the thermal Z → E isomerization of 2˙+–4˙+, which was estimated theoretically (Fig. 2B and Table S7 in the ESI†). The results demonstrated that (Z)-2 shows the lowest oxidation (Eox) and ionization potentials and, therefore, it is more prone to capture a hole from AuNPs. In addition, the isomerization energy barriers for the cationic species are significantly smaller than those of the corresponding neutral species, (Z)-2˙+ presenting a marginally lower value. Based on this evidence, a tentative mechanism is proposed for the large photocatalytic Z → E conversion of 2 (Fig. 3), which consists of: (a) host–guest complex formation onto AuNP@1, (b) light-induced hole transfer to the nearby stilbene molecule, whose isomerization is probably promoted by the concomitant photothermal effect caused owing to the nonnegligible energy barrier computed, and (c) subsequent reduction of (E)-2˙+ at the nanoparticle surface (or by interaction with a free (Z)-2 molecule‡) to render the final neutral product of the reaction. It must be noted that this mechanism is only applicable to low-Eox stilbenes such as 2. However, an analogous photocatalytic scheme evolving through electron injection and isomerization of the resulting radical anion could occur for electron-poor derivatives, which might explain why slightly larger photocatalytic efficiencies were measured for (Z)-4 with respect to (Z)-3 despite presenting lower association constants with β-CD.
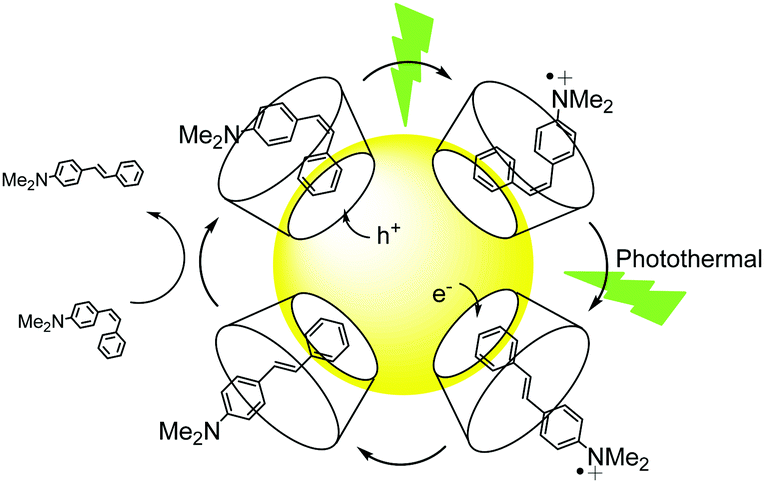 | ||
| Fig. 3 Mechanism proposed for the photocatalytic isomerization of (Z)-2 on AuNP@1. | ||
To corroborate the mechanism proposed in Fig. 3, further experiments were performed with (Z)-4-hydroxystilbene ((Z)-5), another stilbene derivative with a low Eox and a high thermal isomerization barrier that is capable of supramolecularly interacting with β-CD (Tables S1, S7 and S8 in the ESI†). Large photocatalytic Z → E conversion with AuNP@1 was also measured for 5 (88%, Table S4 in the ESI†), thus supporting the reaction scheme devised for electron-rich stilbenes. Actually, nonnegligible (Z)-5 isomerization was observed by the effect of AuNP@1 even in the dark (17%, Table S4 in the ESI†), which hints at a stronger catalyst–substrate interaction in this case that might explain the larger photocatalytic efficiency observed with respect to (Z)-2.
In summary, a new strategy for plasmon-based catalysis has been introduced that relies on the functionalization of noble metal nanoparticles with supramolecular hosts. Although similar approaches have already been used to design drug delivery systems, catalysts for thermal aqueous biphasic reactions and separation agents,15 we prove herein for the first time that our strategy enhances the photocatalytic activity of AuNPs by (a) promoting approximation to the catalyst, (b) ensuring reactant and product renewal, and (c) raising catalyst stability. As a proof of principle, cyclodextrin-coated AuNPs have been shown to photoinduce Z → E isomerization of stilbenes with visible light, obtaining, in some cases, better yields than under direct UV excitation. As revealed by theoretical calculations and electrochemical measurements, such large photocatalytic activity results from the combined action of hole charge injection and photothermal effects caused by AuNPs.
This work was supported by projects CTQ2015-65439-R and CTQ2014-59544-P from the MINECO/FEDER. M. P. and F. P. thank the Universitat Autònoma de Barcelona for a predoctoral PIF fellowship. M. S. gratefully acknowledges support from the ICREA Academia award. X. S.-M. is indebted for the Professor Agregat Serra Húnter position.
Notes and references
- S. Eustis and M. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- (a) H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed; (b) C. M. Cobley, J. Chen, E. C. Cho, L. V. Wang and Y. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC; (c) S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- (a) S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed; (b) J. C. Scaiano and K. Stamplecoskie, J. Phys. Chem. Lett., 2013, 4, 1177–1187 CrossRef CAS PubMed; (c) C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC; (d) G. Baffou and R. Quindant, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC; (e) S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- (a) H. Zhu, X. Ke, X. Yang, S. Sarina and H. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657–9661 CrossRef CAS PubMed; (b) C. J. Bueno-Alejo, C. Fasciani, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Catal. Sci. Technol., 2011, 1, 1506–1511 RSC; (c) Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS PubMed; (d) Z. Liu, Y. Huang, Q. Xiao and H. Zhu, Green Chem., 2016, 18, 817–825 RSC.
- (a) X. Chen, H.-Y. Zhu, J.-C. Zhao, Z.-F. Zheng and X.-P. Gao, Angew. Chem., Int. Ed., 2008, 47, 5353–5356 CrossRef CAS PubMed; (b) G. L. Hallett-Tapley, M. J. Silvero, M. González-Béjar, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, J. Phys. Chem. C, 2011, 115, 10784–10790 CrossRef CAS; (c) P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS; (d) T.-L. Wee, L. C. Schmidt and J. C. Scaiano, J. Phys. Chem. C, 2012, 116, 24373–24379 CrossRef CAS; (e) Y.-F. Huang, M. Zhang, L.-B. Zhao, J.-M. Feng, D.-Y. Wu, B. Ren and Z.-Q. Tian, Angew. Chem., Int. Ed., 2014, 53, 2353–2357 CrossRef CAS PubMed; (f) Q. Xiao, Z. Liu, A. Bo, S. Zavahir, S. Sarina, S. Bottle, J. D. Riches and H. Zhu, J. Am. Chem. Soc., 2015, 137, 1956–1966 CrossRef CAS PubMed.
- (a) J. R. Adleman, D. A. Boyd, D. G. Goodwin and D. Psaltis, Nano Lett., 2009, 9, 4417–4423 CrossRef CAS PubMed; (b) A. B. S. Bakhtiari, D. Hsiao, G. Jin, B. D. Gates and N. R. Branda, Angew. Chem., Int. Ed., 2009, 48, 4166–4169 CrossRef CAS PubMed; (c) C. Fasciani, C. J. Bueno-Alejo, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Org. Lett., 2011, 13, 204–207 CrossRef CAS PubMed; (d) S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS PubMed.
- (a) M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed; (b) R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415–436 CrossRef CAS.
- W. Herrmann, S. Wehrle and G. Wenz, Chem. Commun., 1997, 1709–1710 RSC.
- (a) G. Kistiakowsky and W. R. Smith, J. Am. Chem. Soc., 1934, 56, 638–642 CrossRef CAS; (b) J. Saltiel, S. Ganapathy and C. Werking, J. Phys. Chem., 1987, 91, 2755–2758 CrossRef CAS.
- (a) T. A. Ward, G. Levin and M. Szwarc, J. Am. Chem. Soc., 1975, 96, 258–261 CrossRef; (b) O. Abdul-Rahim, A. N. Simonov, J. F. Boas, T. Ruther, D. J. Collins, P. Perlmutter and A. M. Bond, J. Phys. Chem. B, 2014, 118, 3183–3191 CrossRef CAS PubMed.
- G. L. Hallett-Tapley, C. D'Alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Chem. Commun., 2013, 49, 10075 RSC.
- K. H. Poon and Y. Cheng, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60, 211–222 CrossRef CAS.
- (a) C.-K. Lim, X. Li, Y. Li, K. L. M. Drew, J. P. Palafox-Hernandez, Z. Tang, A. Baev, A. N. Kuzmin, M. R. Knecht, T. R. Walsh, M. T. Swihart, H. Ågren and P. N. Prasad, Nanoscale, 2016, 8, 4194–4202 RSC; (b) T. Miyazawa, S. Koshihara, Y. Segawa and M. Kira, Chem. Lett., 1995, 217–218 CrossRef.
- (a) C. Park, H. Youn, H. Kim, T. Noh, Y. H. Kook, E. T. Oh, H. J. Park and C. Kim, J. Mater. Chem., 2009, 19, 2310–2315 RSC; (b) J. Alvarez, J. Liu, E. Román and A. E. Kaifer, Chem. Commun., 2000, 1151–1152 RSC; (c) Y. Liu, Y.-W. Yang and Y. Chen, Chem. Commun., 2005, 4208–4210 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Further details on the experimental and computational. See DOI: 10.1039/c6cc09600j |
| ‡ Since (Z)-2 has a slightly larger oxidation potential than (E)-2 (see Table S8 in the ESI†), this process should be however thermodynamically disfavored. |
| This journal is © The Royal Society of Chemistry 2017 |