
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of fully arylated (hetero)arenes
Shin
Suzuki
a and
Junichiro
Yamaguchi
 *b
*b
aGraduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan
bDepartment of Applied Chemistry, Waseda University, 3-4-1 Ohkubo, Shinjuku, Tokyo 169-8555, Japan. E-mail: junyamaguchi@waseda.jp
First published on 22nd December 2016
Abstract
Multiply arylated arenes are privileged structures with highly useful functions and fascinating optoelectronic and biological properties. This feature article reports the synthesis of fully arylated (hetero)arenes bearing more than two different aryl substituents and categorizes this emerging topic by the type of (hetero)arene core and the type of chemistry employed to install the (hetero)aryl substituents.
 Shin Suzuki | Shin Suzuki was born in Mie, Japan, in 1990. He obtained a Masters degree in Chemistry from Nagoya University in 2015. Currently, he is a postgraduate student in the group of Kenichiro Itami at Nagoya University, and working with Junichiro Yamaguchi as his co-supervisor focusing on the programmed synthesis of multiply arylated aromatic molecules. |
 Junichiro Yamaguchi | Junichiro Yamaguchi was born in Tokyo, Japan, in 1979. He received his PhD in 2007 from the Tokyo University of Science under the supervision of Prof. Yujiro Hayashi. From 2007 to 2008, he was a postdoctoral fellow in the group of Prof. Phil S. Baran at The Scripps Research Institute (JSPS postdoctoral fellowship for research abroad). In 2008 he became an Assistant Professor at Nagoya University working with Prof. Kenichiro Itami and was promoted to Associate Professor in 2012. He then moved to Waseda University as an Associate Professor (principal investigator) in 2016. His research interests include the total synthesis of natural products and the innovation of synthetic methods. |
Introduction
Benzene, pyridine, thiophene, and other unsaturated ring structures are comprehensively called (hetero)arenes, which represent privileged structural motifs in functional molecules. In particular, structures that have many (hetero)arenes bonded together, i.e., multiply arylated (hetero)arenes, have often been found in natural products, pharmaceuticals and functional organic materials (representative examples are shown in Fig. 1A).1 For example, 1,3-bis(N-carbazolyl)benzene (mCP: 1) and diaryloxadiazole (PBD: 2) have been often seen in the field of organic light-emitting diode (OLED) materials. Oligothiophene (DH-4T: 3) has also displayed activity as a p-type semiconductor. Moreover, widely prescribed pharmaceuticals such as Arcoxia (etoricoxib: 4) and Lipitor (atorvastatin: 5) consist of three aromatic rings as their core, and natural products such as (−)-telomestatin (6) has a unique cyclic oligooxazole structure exhibiting telomerase inhibitor activity. Typically, such molecules have more than two different aryl substituents in order to tune their molecular function. In recent decades, many synthesis methods of multiply arylated (hetero)arenes have been reported.2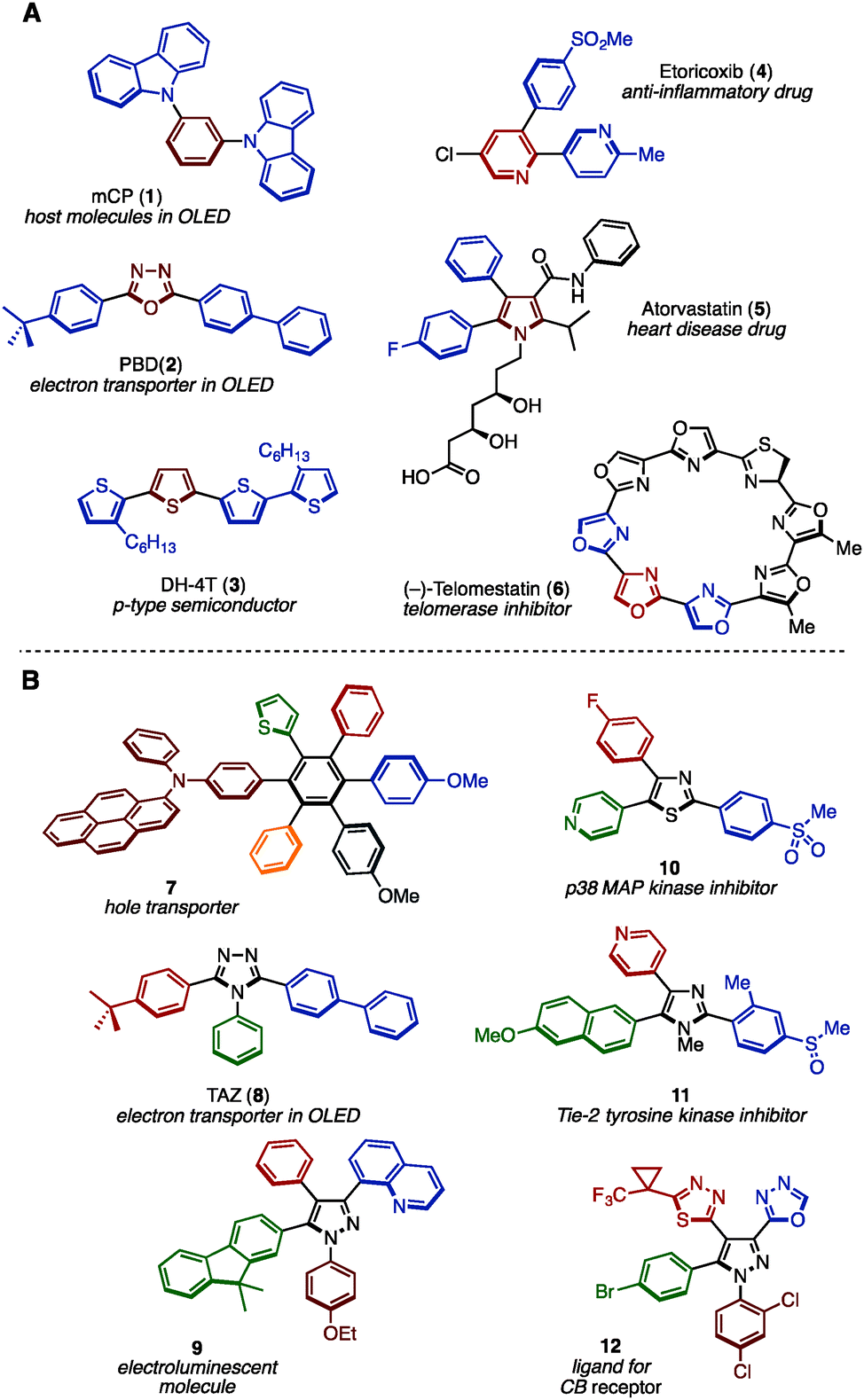 | ||
| Fig. 1 (A) Widely used functional multiply arylated arenes and (B) examples of fully arylated arenes with different aryl groups. | ||
As a subclass of multiply arylated (hetero)arenes, fully arylated (hetero)arenes have also flourished as a unique structural class in functional organic materials and biologically active compounds (representative molecules are shown in Fig. 1B).3 For example, hexaarylbenzene 7 functions as a hole transporter for solar cells. Triaryltriazole, TAZ (8), is known as an electron transporter in OLEDs, and tetraarylpyrazole 9 has been reported as an electroluminescent molecule. Triarylthiazole 10 as well as triarylimidazole 11 act as kinase inhibitors, and triarylpyrazole 12 has been reported as a ligand for the cannabinoid (CB) receptor. Despite the successful application of fully arylated (hetero)arenes with different aryl substituents, the synthesis of such (hetero)arenes has not been explored compared to partially arylated arenes due to the difficulty in synthesizing sterically hindered and highly unsymmetrical aromatic cores.
Despite these synthetic challenges, several compounds have already been utilized in the fields of materials science and biological science, indicating the possibility of fully arylated (hetero)arenes as widely used functional molecules. Therefore, general synthetic methods toward such molecules have recently been developed for the discovery of hitherto unknown functional molecules.
This article introduces recent efforts (made for the past fifteen years with the most emphasis on the past ten) toward fully (hetero)arylated (hetero)arenes bearing more than two different (hetero)aryl substituents, focusing on the synthetic methods used to generate such molecules. We categorized this emerging topic by the type of (hetero)arene core and the type of chemistry employed (mainly, cyclization, cross-coupling, and C–H arylation) to install the (hetero)aryl substituents.
Tetraarylpyrroles, furans, and thiophenes
Cyclization
The Paal–Knorr synthesis is one of the most reliable methods to construct 5-membered aromatic compounds such as pyrroles, furans and thiophenes from 1,4-diketones.4 Recently, using this classical approach, several research groups reported the synthesis of tetraarylpyrroles, tetraarylfurans, and tetraarylthiophenes with more than two different aryl groups (Scheme 1). To synthesize the 1,4-diketone precursor, common strategies involve the oxidative homocoupling (or heterocoupling) of deoxybenzoin derivatives 13 using Cu(OAc)25a,d or I25b as an oxidant or AgF5c as a catalyst to provide tetraarylated 1,4-diketones 14 with two or more different aryl groups. For example, in 2015, Wang and coworkers demonstrated the cross-coupling reaction of 13a (e.g., Ar1 = p-MeOC6H4, Ar2 = p-MeC6H4) and 13b (e.g., Ar3 = p-FC6H4, Ar4 = C6H5) using a Ag catalytic system to synthesize 1,4-diketones 14 bearing four different aryl groups.5c The subsequent condensation of 14 with ammonium acetate gave the corresponding tetraarylpyrrole 15. Treatment of 14 with p-toluenesulfonic acid (TsOH) or Lawesson's reagent (18) also provided tetraarylfurans 16 or tetraarylthiophenes 17, respectively.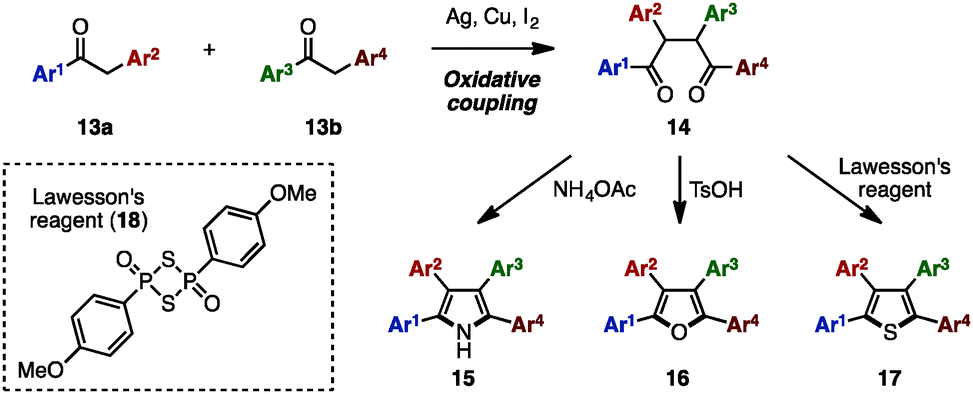 | ||
| Scheme 1 Paal–Knorr synthesis of pyrroles, furans and thiophenes. | ||
In 2007, Opatz and coworkers synthesized tetraarylpyrrole 15a with two different aryl groups by a formal cycloaddition of α-(alkylideneamino)nitrile 19 and nitroolefin 20 with concomitant elimination of HCN and HNO2, albeit in low yields (Scheme 2).6 Although their protocol can potentially provide tetrasubstituted pyrroles with four different substituents, only one example was reported for tetraarylpyrroles.
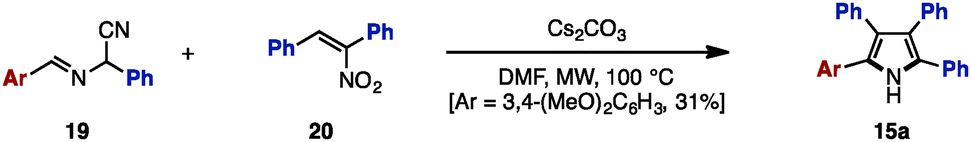 | ||
| Scheme 2 Cycloaddition from (alkylideneamino)nitriles and nitroolefins. | ||
In 2016, Lei and coworkers reported a cross-dehydrative aromatization for the synthesis of tetraarylpyrroles 15 between deoxybenzoin 13 and benzoin derivatives 21 (Scheme 3).7 A benzoin bearing the same aryl groups (21b: Ar3 = Ar4) reacted with 13 in the presence of ammonium acetate in acetic acid, giving tetraarylpyrroles such as 15b with up to three different aryl groups as a single isomer. When benzoins with different aryl groups (21c: Ar3 ≠ Ar4) were employed, tetraarylpyrroles with four different aryl groups were obtained with two regioisomers (15c and 15c′) due to the tautomeric scrambling of the substituents on the benzoin.
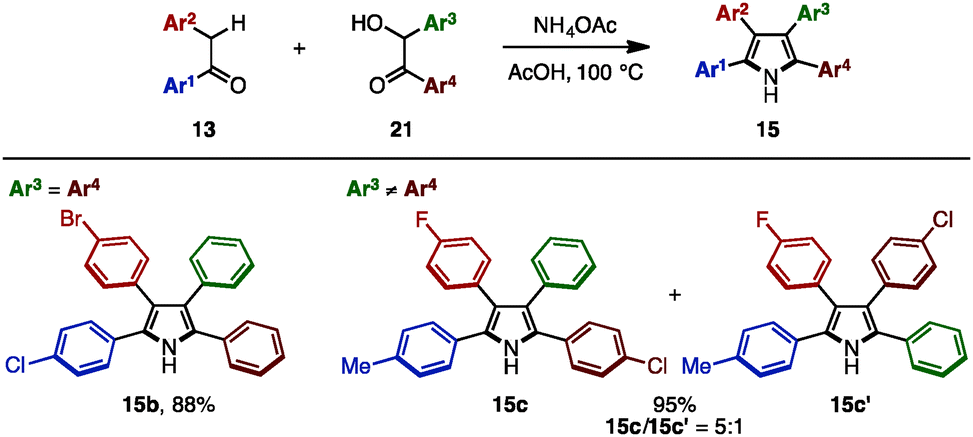 | ||
| Scheme 3 Dehydrative synthesis of tetraarylpyrroles. | ||
Cross-coupling
Transition-metal catalyzed cross-coupling reaction is one of the most reliable methods to install aryl groups onto aromatic molecules, in which the preparation of halogenated and metalated arenes is required prior to the cross-coupling step. Between 2007 and 2011, Langer and coworkers reported multiple cross-coupling reactions of N-methyltetrabromopyrrole (22) and tetrabromothiophene (23), giving N-methyltetraarylpyrroles and tetraarylthiophenes, respectively (Scheme 4A).8 The treatment of 22 or 23 with 5.0 equivalents of arylboronic acid provided N-methyltetraarylpyrroles (methyl-15) and tetraarylthiophenes 17 with a single type of aryl group. By harnessing the different reactivities of the carbon–halogen bonds at the C2- and C3-positions on the pyrrole and thiophene, the synthesis of methyl-15 and 17 bearing two different aryl groups was also achieved through sequential cross-coupling reactions.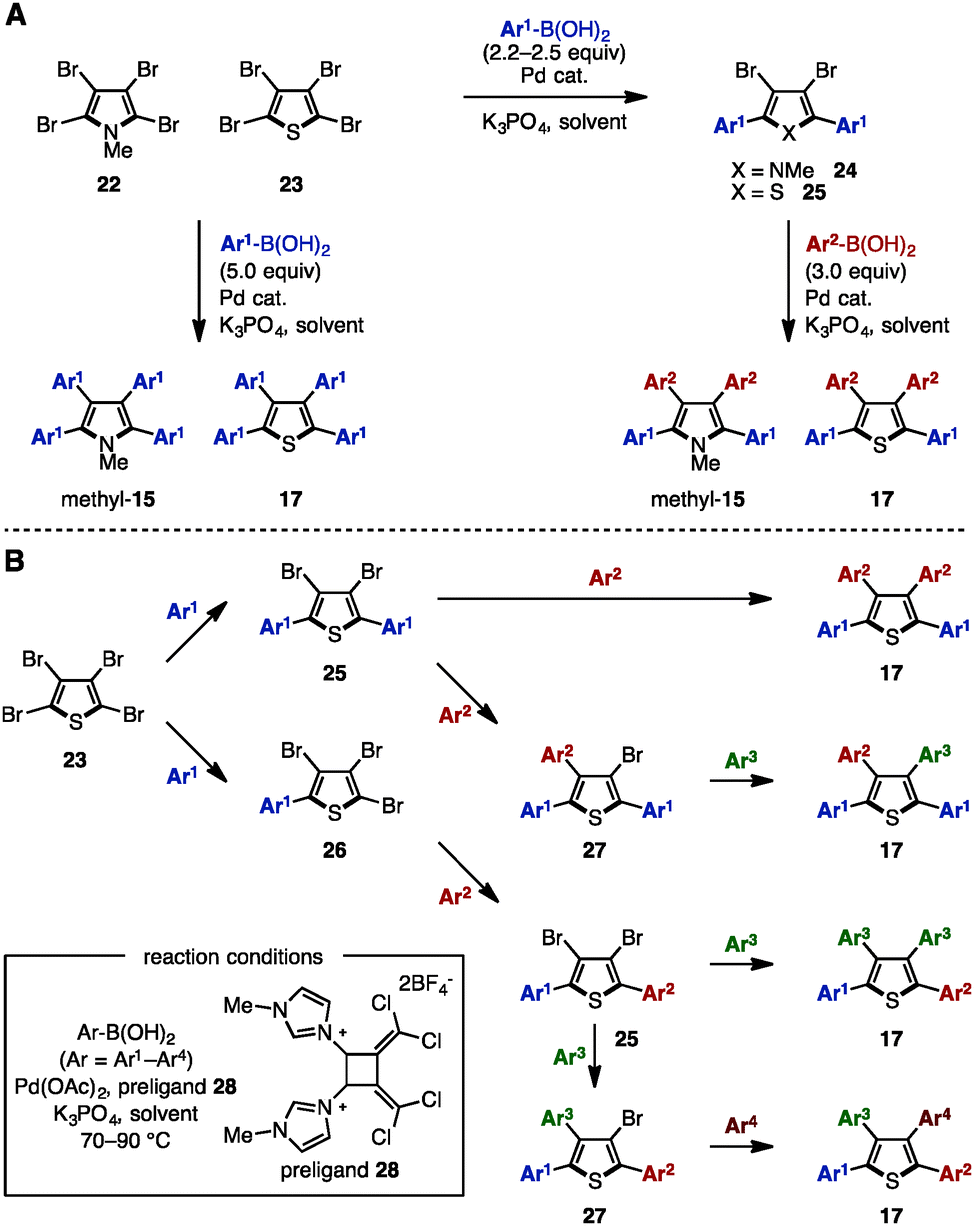 | ||
| Scheme 4 (A) Suzuki–Miyaura cross-coupling of tetrabromopyrroles and thiophenes and (B) sequential Suzuki–Miyaura cross-coupling catalyzed by a Pd-imidazolium salt. | ||
In 2011, Schmidt and coworkers synthesized pre-ligand 28 and applied it in a Pd-catalyzed system to the sequential synthesis of arylated thiophenes 17 bearing up to four different aryl substituents (Scheme 4B).9 Starting from tetrabromothiophene (23), nine different substitution patterns of arylated thiophenes were synthesized by Suzuki–Miyaura coupling under a single set of catalytic conditions (and only changing the reaction temperature and the number of equivalents of arylboronic acid when needed). Although differences in steric bulk must be present in Ar1 (p-tolyl) and Ar2 (o-tolyl) in order to introduce the Ar3 group site-selectively (reaction from compound 25 to 17), this protocol can provide tetraarylthiophenes with four different aryl groups.
In 2011, Yamamoto and coworkers reported an electrophilic iodocyclization of propargyl alcohols for the synthesis of dihaloheterocycles (Scheme 5).10 Treatment of propargyl alcohol 29a with iodine monobromide (IBr) afforded 3-bromo-4-iododihydrothiophene 30. After DDQ-mediated oxidation of 30, subsequent iodo-selective Suzuki–Miyaura cross-coupling with an arylboronic acid furnished triarylthiophene 27. Lastly, Suzuki–Miyaura coupling of 27 and another type of arylboronic acid gave tetraarylthiophene 17 with four different aryl groups. When 4-hydroxy-1,4-diaryl-but-2-yn-1-one 29b was employed instead in this iodocyclization procedure, it furnished 3-bromo-4-iodo-2,5-diarylfuran 31, which can be a precursor for tetraarylfurans with four different aryl substituents.
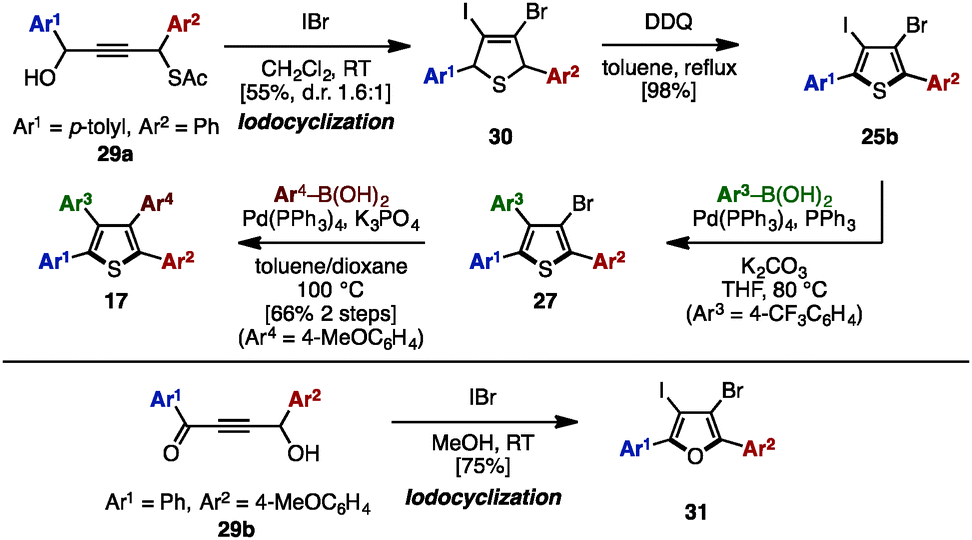 | ||
| Scheme 5 Electrophilic iodocyclization of propargyl alcohols. | ||
In 2015 and 2016, Nishikata and coworkers developed a Cu-catalyzed formal [3+2] cycloaddition for the synthesis of tetraarylfurans 16 and tetraarylthiophenes 17 with four different aryl groups (Scheme 6).11 The [3+2] cycloaddition of styrene derivatives 32 with 2-bromo ketoesters 33 in the presence of a Cu salt, tris(2-pyridylmethyl)amine (TPMA) and diisopropylamine, proceeded, and the subsequent DDQ-mediated oxidation of cycloadduct 34 provided diarylated furans 35 in moderate to high yields as a single isomer. After bromination of 35, Suzuki–Miyaura cross-coupling with arylboronic acids gave triarylfurans 36. Hydrolysis of the methyl ester, followed by decarboxylative coupling with aryl iodides, afforded the corresponding tetraarylfurans 16. In an alternative reaction pathway, intermediate 34 can be first treated with Lawesson's reagent and then oxidized to give thiophenes 37, which could be converted to tetraarylthiophenes 17 in a similar manner.
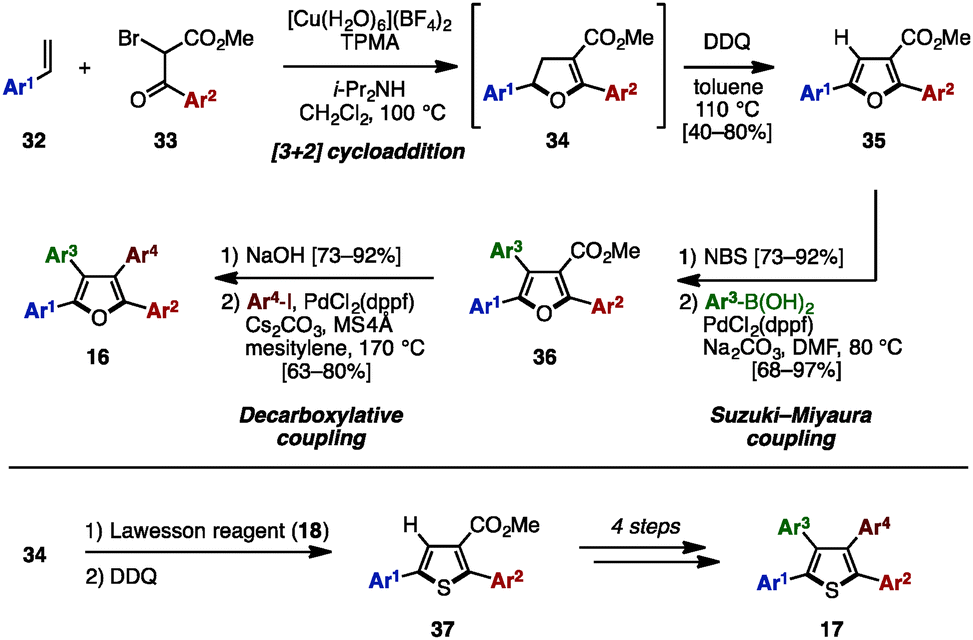 | ||
| Scheme 6 Cu-catalyzed formal [3+2] cycloaddition. | ||
C–H arylation
C–H arylation of (hetero)arenes not only enables the shortening of synthetic steps compared to typical cross-coupling reactions, but can also allow control of the position of aryl substituents at will.12 In 2008, Miura and coworkers developed multiple arylation of 3-thiophene- (38) and 3-furancarboxylic acid (39), consisting of C–H arylation and decarboxylative arylation reactions (Scheme 7).13 Treatment of 38 or 39 with excess aryl bromide in the presence of Pd(OAc)2 and PCy3 afforded tetraarylthiophenes 17 or tetraarylfurans 16 in moderate to high yields, although a small amount of triarylated isomer was also detected. When ethyl-3-thiophenecarboxylate (40) was selected as a substrate, 2,5-diarylation proceeded under Pd catalysis [Pd(OAc)2/Johnphos] to provide 2,5-diarylthiophenes 41 in high yields. After hydrolysis of the ester moiety of 41, the resulting carboxylic acids 42 were subjected to a Pd-catalyzed C–H arylation and a decarboxylative arylation with aryl halides, providing tetraarylthiophenes 17 with two different aryl groups.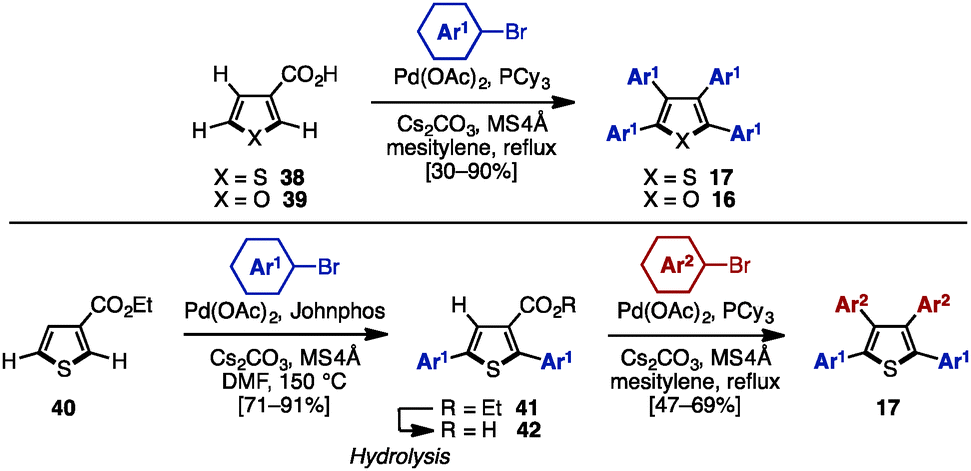 | ||
| Scheme 7 Decarboxylative coupling of 3-thiophene- and 3-furancarboxylic acid. | ||
In 2009, Itami and coworkers demonstrated a programmed synthesis of tetraarylthiophenes 17 with four different aryl substituents by using a sequential regioselective C–H arylation strategy starting with 3-methoxythiophene (43: Scheme 8).14 The synthesis commenced with C2-arylation of 43 with aryl iodides in the presence of RhCl(CO){P[OCH(CF3)2]3}2,15 giving 2-aryl-3-methoxythiophenes 44 with virtually complete regioselectivity. A C4-selective arylation of 44 with aryl iodides was catalyzed by PdCl2/P[OCH(CF3)2]3 to afford 2,4-diaryl-3-methoxythiophenes 45 with high β-selectivity.16 Treatment of 45 with aryl iodides using a PdCl2/2,2′-bipy catalyst then provided 2,4,5-triaryl-3-methoxythiophenes 46.16 After demethylation of 46, followed by triflation of the resulting alcohol, triflates 47 were coupled with arylboronic acids in the presence of Pd(PPh3)4 to produce tetraarylthiophenes 17 with virtually complete isomeric purities.
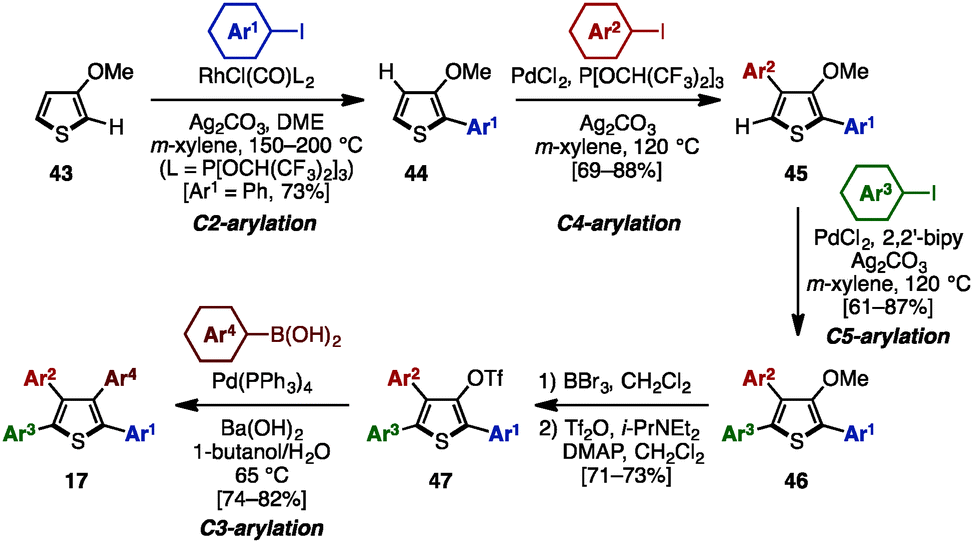 | ||
| Scheme 8 Programmed synthesis of tetraarylthiophenes. | ||
Triarylated 1,2-azoles and 1,3-azoles
Cyclization
Between 2013 and 2015, in order to synthesize N-arylmethyltriarylimidazoles 50 and triaryloxazoles 51, several research groups reported an oxidative cyclization between benzil derivatives 48 and benzylamines 49 by using metal salts such as NiCl2·6H2O,17a CuI,17b,d and Ag2CO317c (Scheme 9A). In addition, triaryloxazoles 51 were synthesized by cyclization of benzoates 53 (which can be prepared by acylation of benzoins 21 with aroyl chlorides 52) in the presence of a NH3 source (Scheme 9B).18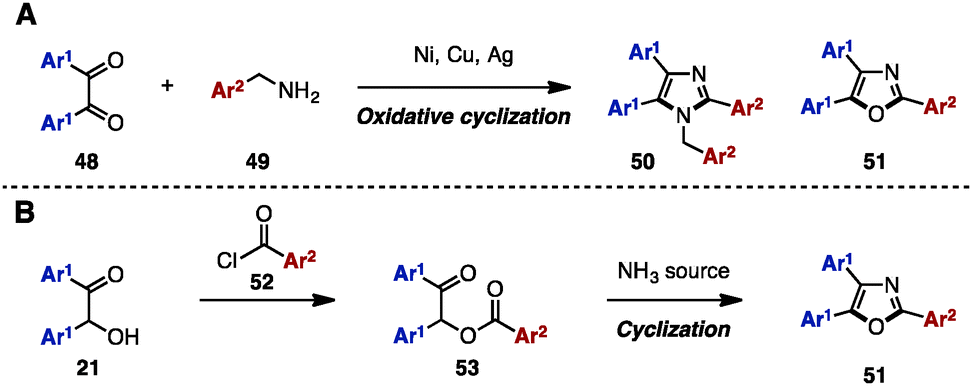 | ||
| Scheme 9 Imidazole and oxazole synthesis from benzil and benzoin derivatives. | ||
In 2008, Opatz and a coworker reported the modular synthesis of tetrasubstituted imidazoles and trisubstituted oxazoles by cross-coupling N-acylimines 54 and α-aminonitriles 55 (Scheme 10).19 The synthesis began with a 1,2-addition of deprotonated α-aminonitriles 56 onto N-acylimines 54, affording the corresponding adducts 56. Treatment of 56 with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under refluxing toluene furnished α-acylaminoimines 57, which can spontaneously cyclize to imidazoles 58 depending on the substitution pattern at the C5 position (Ar3). Bulky substituents at the C5 position promoted the spontaneous cyclization to form 58, but in other cases, 57 could be converted to imidazoles 58 by treatment with PCl5. Hydrolysis of imines 57 under acidic conditions provided the corresponding ketones, which were dehydrated with PCl5 to afford triaryloxazoles 51 with three different aryl groups.
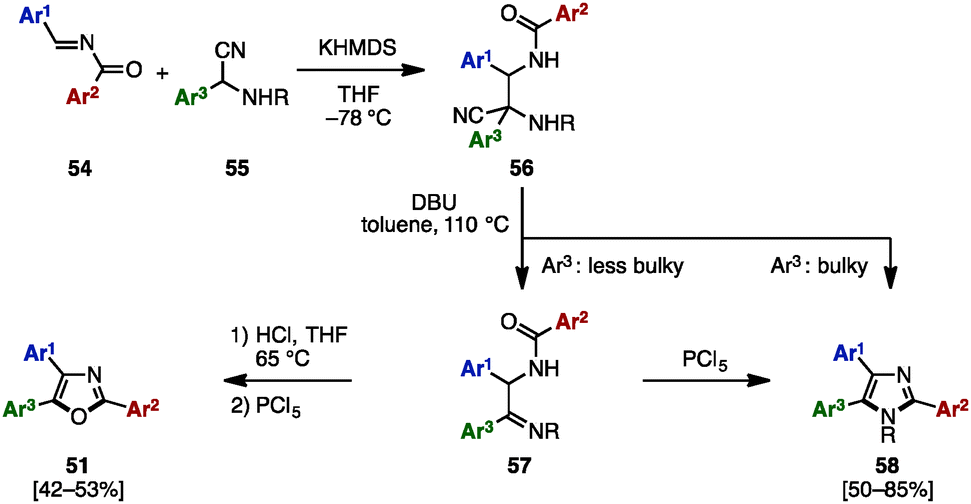 | ||
| Scheme 10 Modular synthesis of triarylimidazoles and triaryloxazoles. | ||
The Bredereck synthesis is one of the fundamental methods to construct substituted oxazoles and thiazoles.20 Using this approach, in 2014, Bailey and a coworker reported a silver-promoted oxazole synthesis with α-bromoketones 58 and aryl amides 59, giving triaryloxazoles 51 in moderate yields (Scheme 11).21 Cyclization with aryl thioamide 60 instead of 59 smoothly proceeded even in the absence of silver salt to form triarylthiazoles 61.
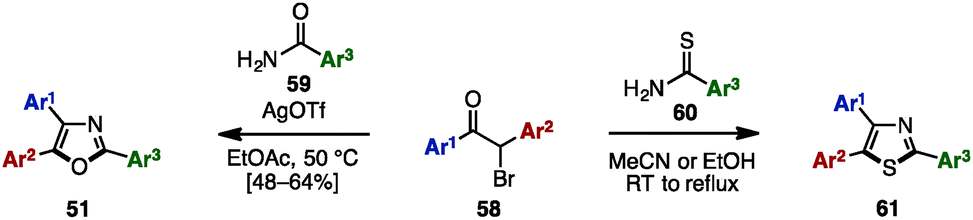 | ||
| Scheme 11 Bredereck synthesis of oxazoles and thiazoles. | ||
In 2014, Lin and coworkers applied an intramolecular Wittig-type reaction for the synthesis of trisubstituted oxazoles from N-acylimines 54 (Scheme 12).22 Treatment of 54 with acyl chlorides 52 in the presence of P(n-Bu)3 (62) and Et3N provided triaryloxazoles 51 with three different aryl groups in moderate to excellent yields. The mechanism of this reaction was proposed to be an intramolecular Wittig-type reaction of presumable phosphorus ylides 63, which were formed by 1,4-addition of P(n-Bu)3 (62) to 54 and O-acylation with 52.
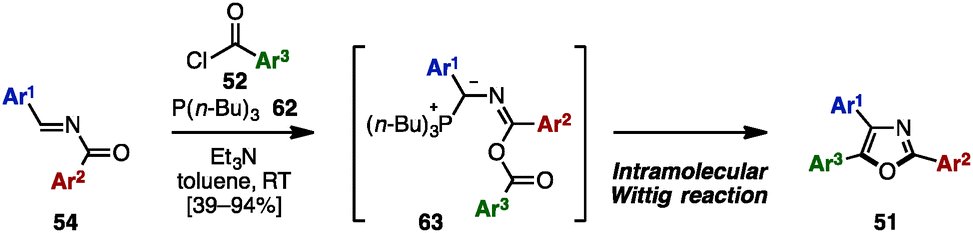 | ||
| Scheme 12 Triaryloxazole synthesis via the intramolecular Wittig reaction. | ||
Recently, visible-light photoredox catalysis has attracted significant attention as a green and sustainable synthetic method to make substituted heterocycles under mild reaction conditions.23 In 2015, Xiao and coworkers disclosed the synthesis of trisubstituted oxazoles including triaryloxazoles from 2H-azirines 64 and aryl aldehydes 65 by using 9-mesityl-10-methylacridinium perchlorate 66 as the photoredox catalyst (Scheme 13A).24a To this end, a formal [3+2] cycloaddition of 64 and 65 proceeded at room temperature in the presence of catalytic 66 under irradiation with blue LED light to provide 2,5-dihydrooxazoles 67. Subsequent DDQ-mediated oxidation provided the corresponding triaryloxazoles 51 with three different aryl groups in a one-pot operation, which could be utilized for the synthesis of cyclooxygenase-2 inhibitor 51a. Furthermore, in 2016, Cho and coworkers synthesized triaryloxazoles 51b and 51c from α-bromoketone 58a and benzylamine derivatives 49 by means of a Ru-photoredox catalyst under blue LED light irradiation (Scheme 13B).24b
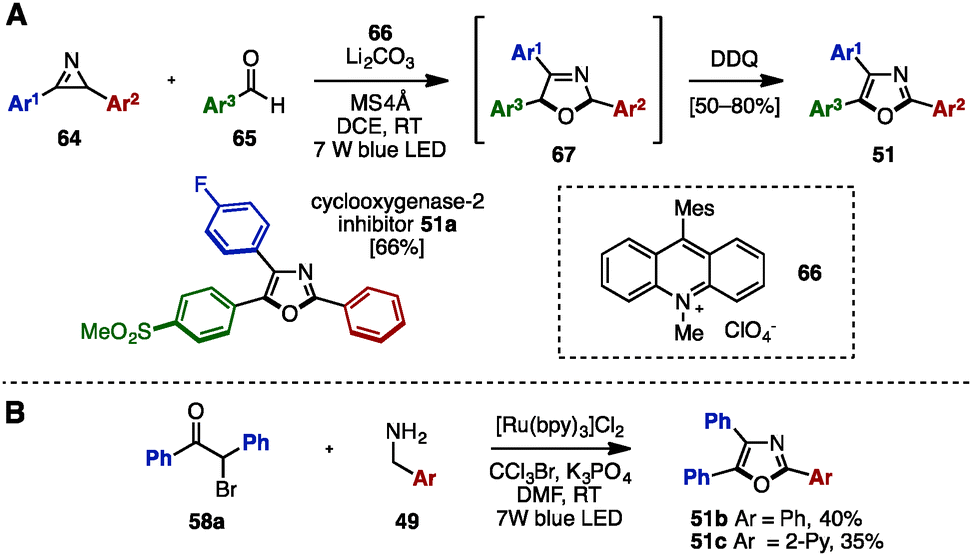 | ||
| Scheme 13 Triaryloxazole synthesis enabled by photoredox catalysis. | ||
To construct an isoxazole core, 1,3-dipolar cycloaddition of aryl nitrile oxides with alkenes or alkynes has been used.25 In 2013, Shetty and coworkers reported the synthesis of 3,4,5-triarylisoxazoles 70 by a 1,3-dipolar cyclization of nitrile oxide (prepared in situ from oxime 68) and styrene to give isoxazole 69, followed by bromination and Suzuki–Miyaura cross-coupling (Scheme 14A).26a In 2005, the group of Denmark synthesized triarylisoxazoles 70 by performing a 1,3-dipolar cyclization of phenylethynyl silyl ether 71 with 72 to give 73, followed by Hiyama cross-coupling with aryl iodides (Scheme 14B).26b Elaborating this further, in 2011, Vasam, Vadde and coworkers achieved an NHC-catalyzed regioselective 1,3-dipolar cycloaddition of diarylacetylenes 74 and aryl nitrile N-oxides 75 to afford triarylisoxazoles 70 regioselectively (Scheme 14C).26c
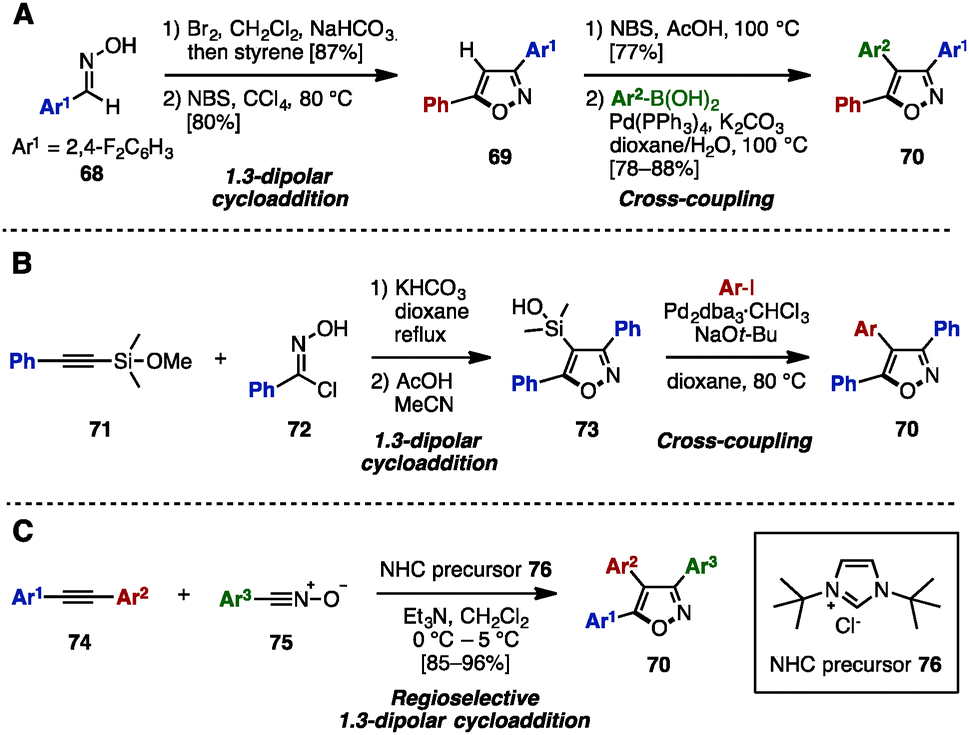 | ||
| Scheme 14 1,3-Dipolar cycloaddition of nitrile oxides with alkenes or alkynes. | ||
Cross-coupling
As mentioned above, the cross-coupling reaction is a powerful method to construct aryl–aryl frameworks. However, examples of multi-substituted azoles synthesized by cross-coupling reactions are quite rare,27 due to the difficulty in accessing metalated azoles and halogenated azoles (they are less stable compared to those of pyrroles and thiophenes). One of few examples was reported in 2002, when Hodgetts and coworkers synthesized triaryloxazoles from 2-chlorooxazole-4-carboxylate (78) by using a sequence of regiocontrolled halogenation and Pd-catalyzed Suzuki–Miyaura coupling (Scheme 15).27b Intermediate 78 was prepared from 2-aminooxazole 77 by treatment with t-BuONO and CuCl2. 78 was then coupled with phenylboronic acid under Pd catalysis, giving 2-phenyloxazole 79. Treatment of 79 with N-bromosuccinimide (NBS) provided a C5-brominated oxazole, which was subjected to a second cross-coupling with phenylboronic acid to afford 2,5-diphenyloxazole 80. After hydrolysis of the ester in 80, the resulting carboxylic acid was converted to bromooxazole 81via a Hunsdiecker reaction. Lastly, Negishi coupling of 81 with 2-pyridylzinc bromide gave triaryloxazole 51d.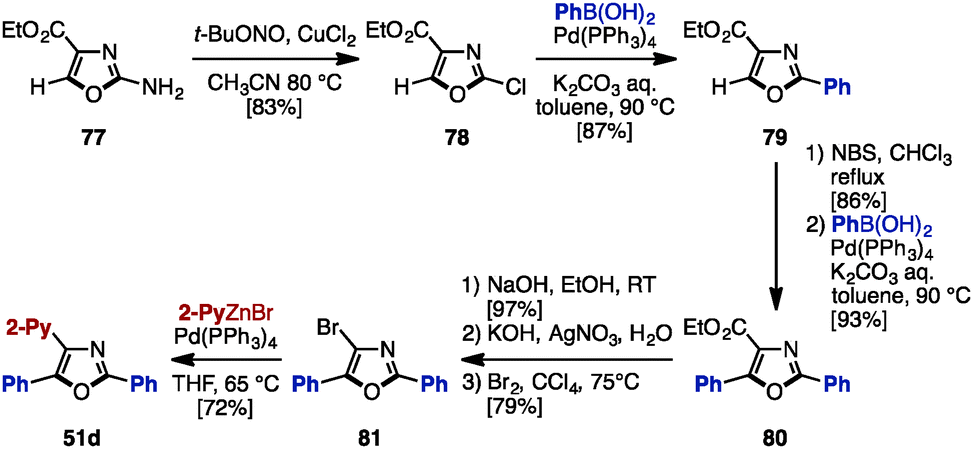 | ||
| Scheme 15 Sequential cross-coupling of ethyl 2-chlorooxazole-4-carboxylate. | ||
In 2013, Knochel and coworkers achieved an exhaustive functionalization of imidazole scaffolds by a combination of chemoselective direct metalation and sulfoxide/magnesium exchange (Scheme 16A).28 Imidazole 82 was designed as the key intermediate of this transformation, in which the N,N-dimethylsulfamoyl group worked as an ortho-directing group and the 4-methoxy-3,5-dimethylbenzenesulfinyl (AnS(O)) group enabled direct metalation to the C4-position. After direct metalation, AnS(O) can be replaced by sulfoxide/magnesium exchange. This synthesis began with the selective metalation of 82 at the C4-position by using TMPMgCl·LiCl, followed by transmetalation of Mg with Zn, and Negishi coupling with aryl iodides to give 4-arylimidazoles 83. Treatment of 83 with i-PrMgCl·LiCl promoted a sulfoxide/magnesium exchange, giving the corresponding Mg species. As with the C4-functionalization, a sequence of transmetalation of the resulting Mg species to Zn and then Negishi coupling yielded 4,5-diarylimidazoles 84. Finally, after removal of the TBS group of 84, deprotonation by TMPMgCl·LiCl (which required transmetalation with ZnCl2 before cross-coupling) or by TMP2ZnCl·2MgCl2·2LiCl at the C2 position, followed by Negishi coupling, furnished sulfonated triarylimidazoles 85 with three different aryl groups.
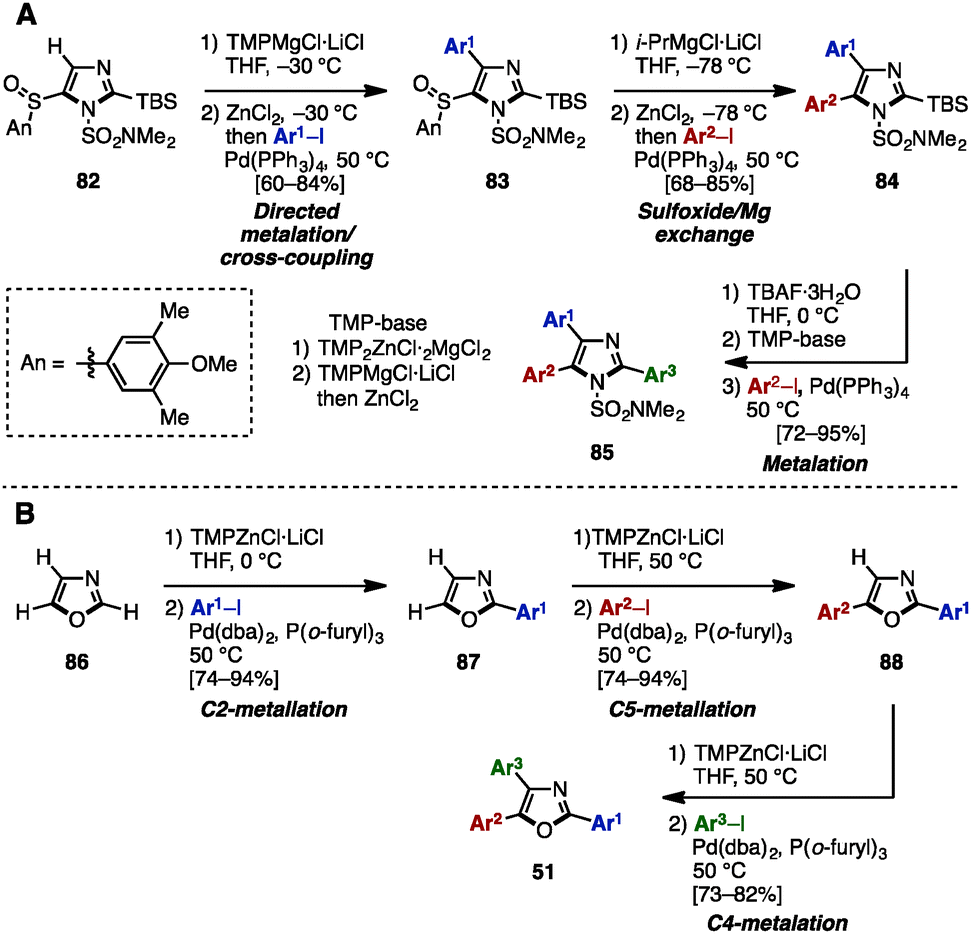 | ||
| Scheme 16 (A) Selective metalation and sulfoxide/magnesium exchange. (B) Regioselective sequential metalation of oxazoles. | ||
In the same year, the same group applied a sequence of regioselective metalations and cross-coupling reactions for the synthesis of triaryloxazoles 51 from simple oxazole (86) (Scheme 16B).29 The use of TMPZnCl·LiCl as a metalation reagent and appropriately controlling the reaction temperature enabled regioselective metalations at the C1-, C4-, and C3-positions of oxazoles to give the corresponding zincated oxazoles. These intermediates were then reacted with aryl iodides in the presence of catalytic Pd(dba)2/P(o-furyl)3 to provide arylated oxazoles. As a result, triaryloxazoles 51 could be synthesized regioselectively from simple oxazole (86) in three metalation/cross-coupling sequences.
In 2008, McLaughlin and coworkers developed a synthesis of 3,4,5-triarylpyrazoles using a switchable metal-directing group [a tetrahydropyran (THP) group], which enabled direct sequential lithiation of the C3- and C5-positions of the pyrazole core (Scheme 17).30 First, THP-protected pyrazole 89 was lithiated by n-BuLi at the C5-position, followed by treatment with B(Oi-Pr)3 and pinacol, providing 5-borylpyrazole 90 regioselectively. Subsequent cross-coupling reaction with aryl bromides proceeded to furnish 5-arylpyrazoles 91. By moving the metal-directing group (THP) from N1 to N2, the site of lithiation on the pyrazole core was shifted from C5 to C3. Taking advantage of this selectivity, a sequence of lithiation and borylation afforded 5-boryl-3-arylpyrazole 92, which was coupled with aryl bromides to give 3,5-diarylpyrazoles 93. Lastly, 3,4,5-triarylpyrazole 94 was synthesized by treatment of 93 with a sequence of bromination, cross-coupling, and removal of the THP group.
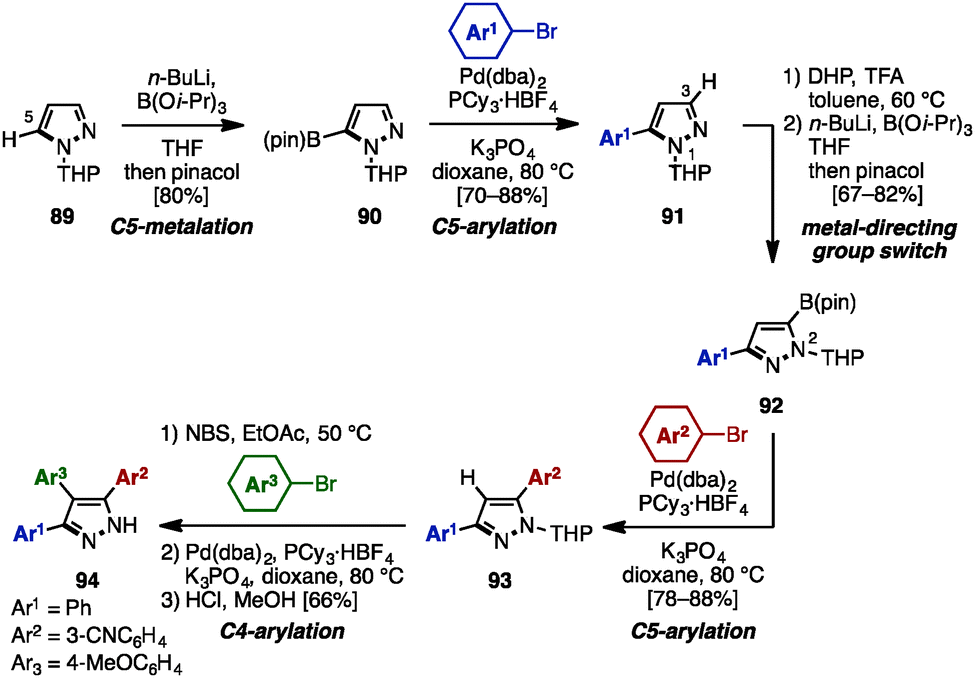 | ||
| Scheme 17 Synthesis of 3,4,5-triarylpyrazole by using a switchable metal-directed group. | ||
C–H arylation
To avoid pre-functionalization and the use of unstable metallo-1,3- and 1,2-azoles, C–H arylation of azoles is one recent solution for the synthesis of multi-arylated azoles. In 2009, Fagnou and coworkers developed regioselective multiple C–H arylations of azole N-oxides (Scheme 18).31 The N-oxide group not only enhanced the reactivity at all positions of azole derivatives, but also enabled a differentiated reactivity of C2 and C5 positions. First, C2-arylation of thiazole N-oxide (95) with aryl iodides proceeded at room temperature in the presence of catalytic Pd(OAc)2, PhDavephos, CuBr, Cs2CO3, and PivOH in toluene to afford 2-arylthiazole N-oxides 96 with virtually complete regioselectivity. The addition of CuBr suppressed the production of a C5/C2 doubly arylated product. Since the C2 position was blocked, thiazole N-oxide 96 underwent a highly selective C5-arylation using catalytic Pd(OAc)2/P(t-Bu)3 and K2CO3 in toluene to provide 2,5-diarylthiazole N-oxides 97. C4-arylation of 97 with aryl bromides proceeded in the presence of Pd(OAc)2, PPh3, and K2CO3 in toluene, giving triarylthiazole N-oxide 98. Finally, thiazole N-oxide 98 could be deoxygenated to triarylthiazole 61 by treatment with Zn powder and aqueous NH4Cl in THF. This protocol is applicable to the synthesis of triarylimidazole N-oxides 101 as well.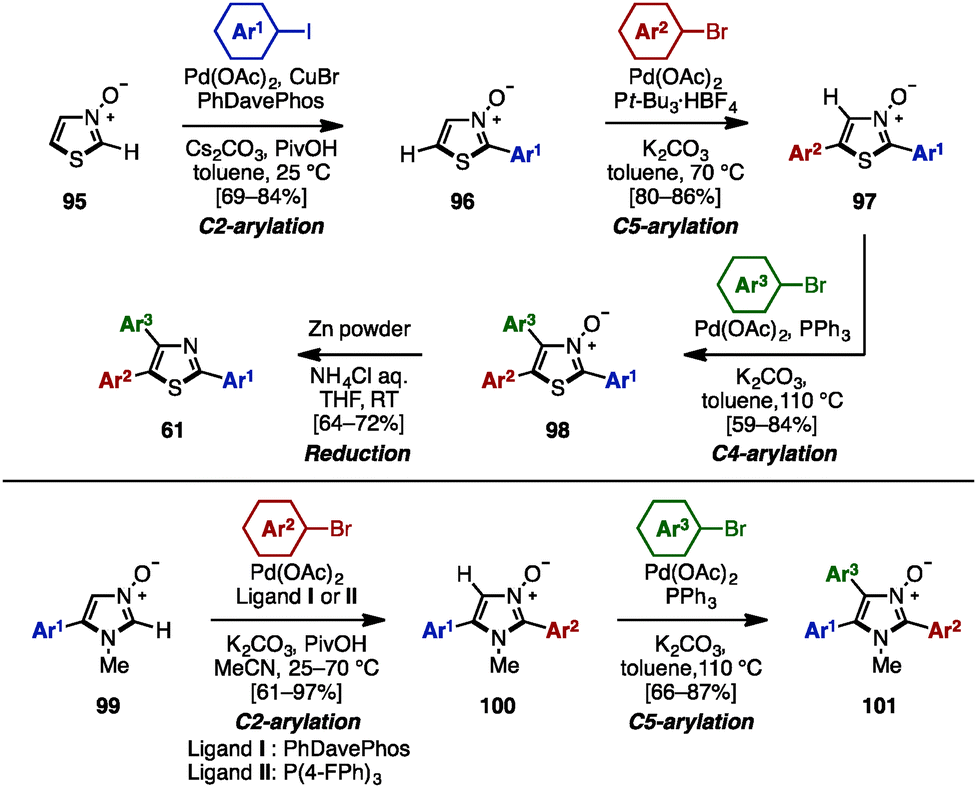 | ||
| Scheme 18 Regioselective sequential C–H arylation of azole N-oxides. | ||
In 2010, Sames and coworkers reported a regioselective sequential C–H arylation of SEM-protected imidazole 102 by using a “SEM-switch” strategy (Scheme 19).32 This strategy transfers a SEM group from the N-1 to the N-3 nitrogen atom and thus enables a switch of the reaction site on imidazole cores. First, C5-arylation of 102 with aryl bromide proceeded in the presence of Pd(OAc)2, P(n-Bu)Ad2, and K2CO3 in dimethylacetamide (DMA) to provide 5-arylimidazoles 103. Subsequently, 103 was coupled with aryl bromides under Pd(OAc)2/P(t-Bu)3 catalysis using NaOt-Bu as a base to furnish 2,5-diarylimidazoles 104 with complete regioselectivity. By means of a SEM-switch or N-alkylation, the reactive site of 104 was shifted to the C5 position of 105a or 105b (previously the C4 position of 102). Finally, C5-arylation of 105a and 105b proceeded under the same reaction conditions as the first arylation step, giving triarylimidazoles 106a and 106b.
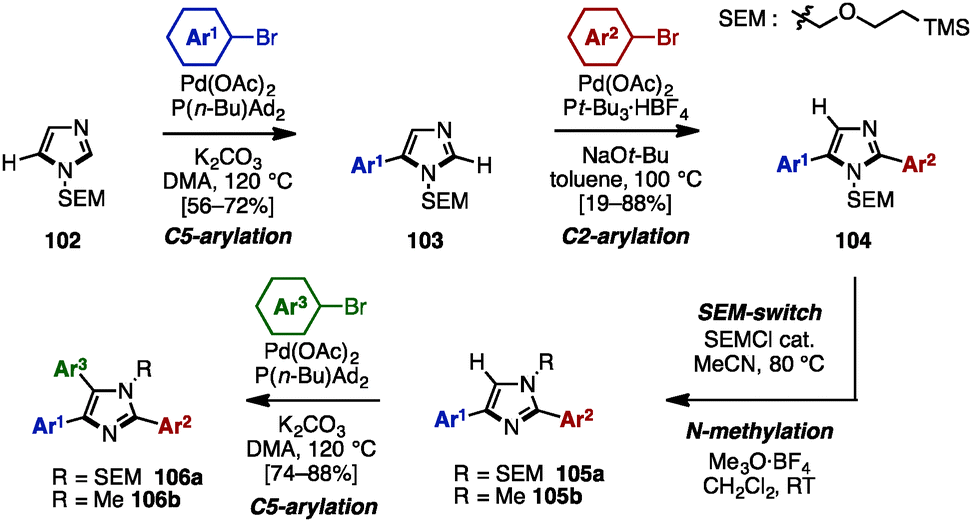 | ||
| Scheme 19 Regioselective sequential C–H arylation of SEM-protected imidazoles. | ||
In 2011, Murai/Shibahara and coworkers reported a multiple regiocontrolled C–H arylation of simple 1,3-azoles such as N-methylimidazole (107), oxazole, and thiazole by [Pd(phen)2](PF6)2 catalyst 108 (Scheme 20A).33 For example, 107 was coupled with aryl iodides in the presence of 108 as a catalyst to afford 5-arylimidazoles 109. Treatment of 109 with aryl iodides and catalyst 108 yielded 2,5-diarylimidazoles 110 with high regioselectivity when galvinoxyl was used as an additive. A subsequent reaction also using Pd catalyst 108 enabled the C4-arylation of 110, providing triarylimidazoles 106b. This three-step sequence was applicable not only to the sequential regioselective triarylation of thiazole, but also to the synthesis of Tie-2 tyrosine kinase inhibitor 11 (see Fig. 1).
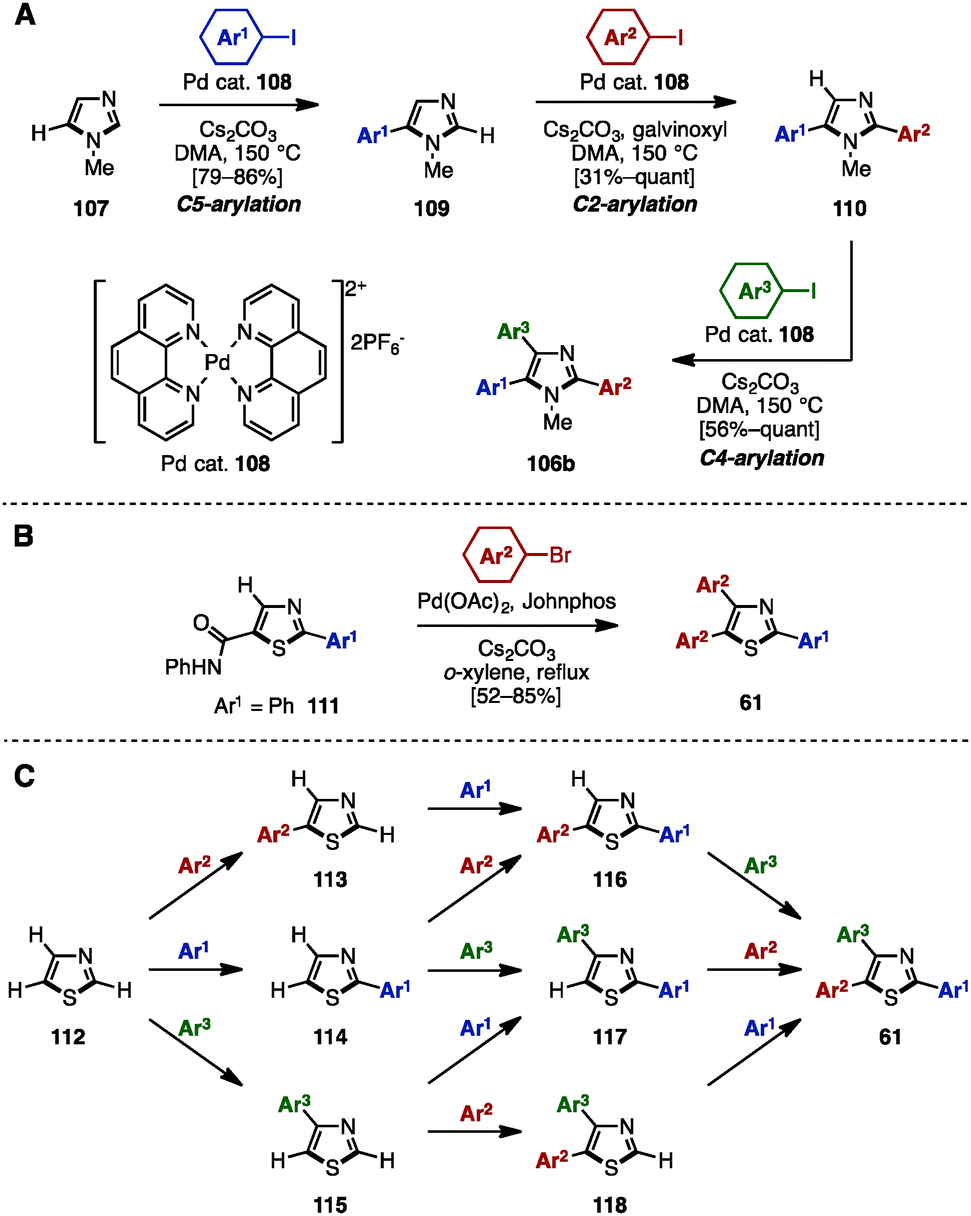 | ||
| Scheme 20 (A) Multiple regioselective direct arylation of simple azoles, (B) C–H arylation of 5-thiazolecarboxanilide and (C) programmed synthesis of arylthiazoles. | ||
In 2003, Miura and coworkers reported the diarylation of 2-phenyl-5-thiazolecarboxyanilide (111) at the C4 and C5 positions with concomitant decarbamoylation (Scheme 20B).34 The reaction of 111 with aryl bromides in the presence of Pd(OAc)2, Johnphos, and Cs2CO3 in refluxing o-xylene afforded the diarylated product, triarylthiazoles 61, with two different aryl groups by decarbamoylation and C–H arylations of thiazoles.
In 2014, Itami and coworkers achieved a programmed synthesis of arylthiazoles via sequential direct C–H arylation reactions (Scheme 20C).35 Although the synthesis of triarylthiazoles from 2-phenylthiazole by C–H arylation was already a known method at the time,36 all possible substitution patterns of arylthiazoles 113–118 and 61 could be synthesized using this synthetic protocol from simple thiazole (112) via 11 distinct routes. Furthermore, this method enabled not only a gram-scale synthesis of triarylthiazole 61 with three different aryl groups, but also the preparation of over 150 different arylthiazoles.
The SEM-switch strategy (see Scheme 19) developed by Sames and coworkers was also applicable to the synthesis of triarylpyrazoles 120 (Scheme 21).37 The synthesis began with a cross-coupling reaction of 4-bromopyrazole 119 with arylboronic acids, giving 4-arylpyrazoles 120. Treatment of 120 with aryl bromides in the presence of Pd(OAc)2, P(n-Bu)Ad2, K2CO3, and PivOH in N,N-dimethylacetamide (DMA) provided 4,5-diarylpyrazoles 121. A SEM-switch or N-methylation of 121 afforded 3,4-diarylpyrazoles 122a or 122b, which were coupled with aryl bromides to furnish triarylpyrazoles 123a or 123b with three different aryl substituents.
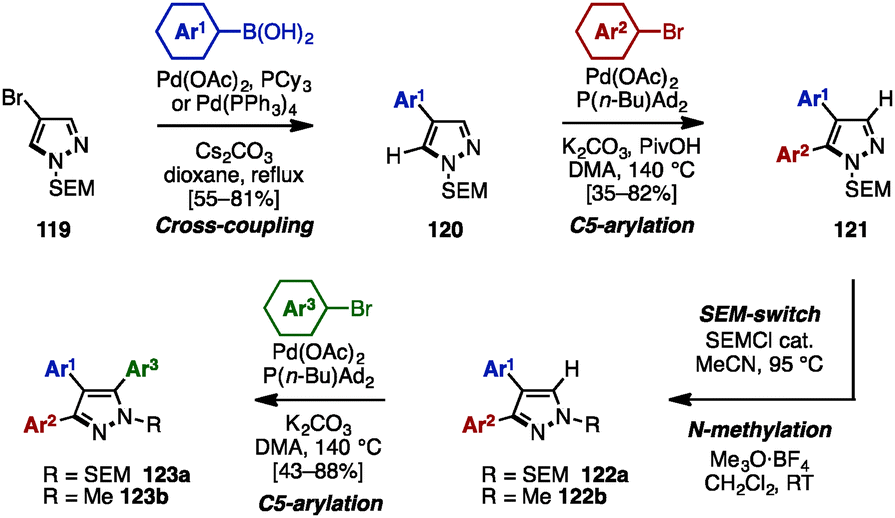 | ||
| Scheme 21 Regioselective sequential C–H arylation of SEM-protected pyrazoles. | ||
In 2015, Fuse and coworkers reported the regioselective synthesis of 1,3,4,5-tetraarylpyrazoles using a sequence of SNAr, cross-coupling, and C–H arylation reactions, in which 3-iodo-1H-pyrazole 124 was selected as a starting material (Scheme 22A).38 Iodopyrazole 124 was a mixture of tautomers 124a and 124b, however, SNAr reaction of 124 with aryl fluorides provided N-arylpyrazole 125 as a single isomer. Subsequent cross-coupling of 125 with arylboronic acids or arylpinacol esters using catalytic Pd2(dba)3/P(t-Bu)3 furnished 3-arylpyrazoles 126. C5-arylation of 126 with aryl bromides occurred in the presence of Pd(OAc)2, PCy3, K2CO3, and PivOH in 1,4-dioxane to afford 1,3,5-triarylpyrazole 127. By changing the ligand and solvent from PCy3 and 1,4-dioxane to P(n-Bu)Ad2 and DMA, C4-arylation of 127 proceeded to afford tetraarylpyrazole 128 with virtually complete isomeric purities.
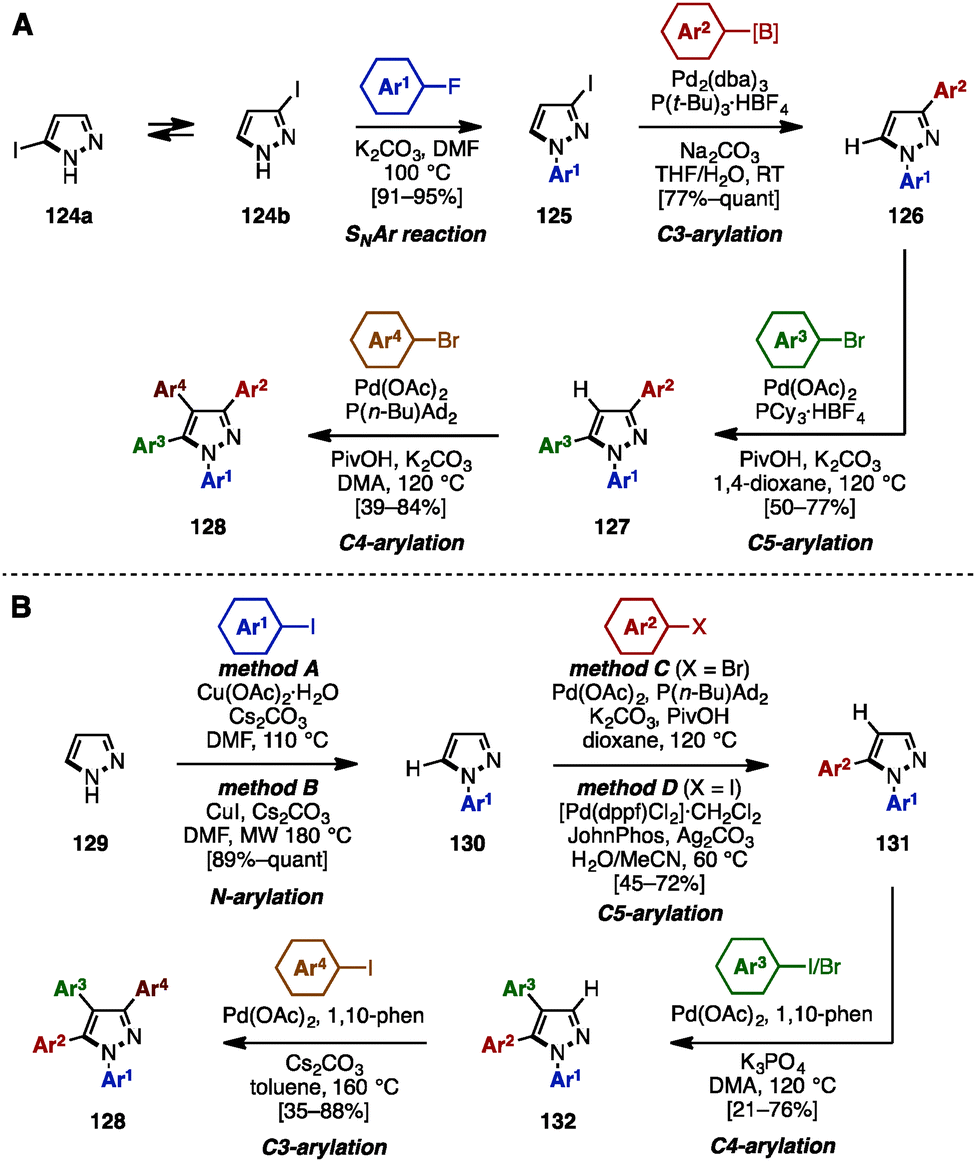 | ||
| Scheme 22 (A) Sequential SNAr/C–H arylation/cross-coupling of iodopyrazoles and (B) four-fold regioselective direct arylation for the synthesis of tetraarylpyrazoles. | ||
In 2016, the same group achieved the synthesis of 1,3,4,5-tetraarylpyrazole 128 by using four-fold regioselective direct arylation reactions of simple pyrazole (129), including N–H arylation and three C–H arylations (Scheme 22B).39 First, pyrazole (129) was subjected to Cu-catalyzed N–H arylation, providing N-arylpyrazoles 130 in excellent yields using a choice of two conditions (method A or B). A regioselective C5-arylation of 130 was then accomplished by selecting the reaction conditions (method C or D) depending on the substituent (Ar2) of the aryl halide component to furnish 1,5-diarylpyrazoles 131 in moderate to good yields. C4-arylation of 131 with aryl iodides or aryl bromides proceeded in the presence of Pd(OAc)2, 1,10-phenanthroline (1,10-phen), and K3PO4 in DMA to afford 1,4,5-triarylpyrazole 132. Finally, the synthesis of 1,3,4,5-tetraarylpyrazoles 128 was achieved by treatment with aryl iodides in a Pd(OAc)2/1,10-phen catalytic system.
In the same year, Fuse/Nakamura and coworkers accomplished the synthesis of triarylisoxazoles 70 by a sequence of cross-coupling and C–H arylation reactions (Scheme 23).40 This synthesis began with a cross-coupling reaction of 4-iodo-3-phenylisoxazole 133 with an arylboronic acid, giving 3,4-diarylisoxazole 134 in excellent yield.
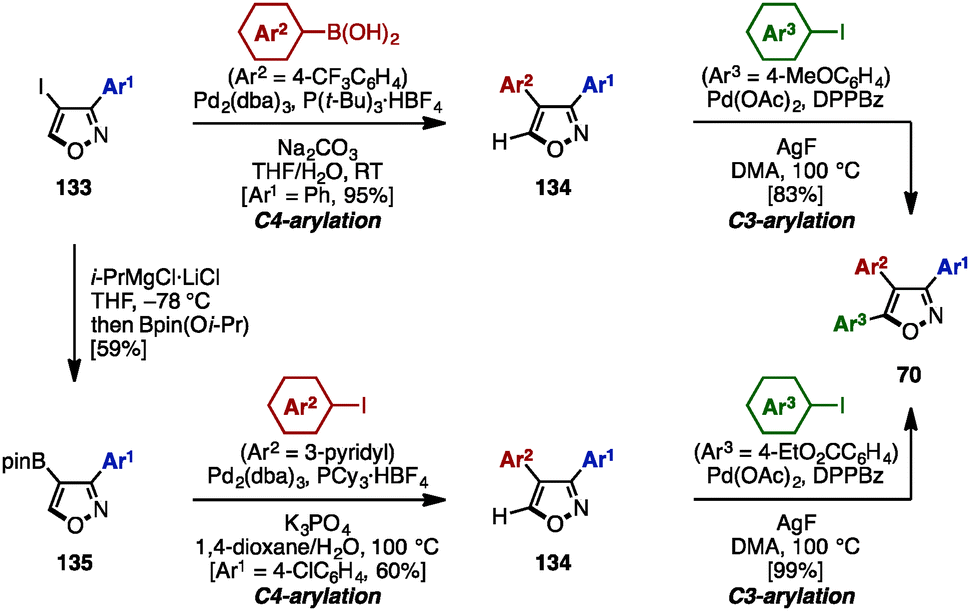 | ||
| Scheme 23 Synthesis of triarylisoxazoles by cross-coupling and C–H arylation reactions. | ||
Subsequent C–H arylation at the C5-position of isoxazole using a procedure developed by Sasai and Takenaka41 furnished triarylisoxazole 70 with three different aryl groups. In an alternative reaction sequence, 4-iodoisoxazole 133 was reacted with “turbo Grignard” reagent (i-PrMgCl·LiCl) in THF at −78 °C to produce a 4-isoxazolyl anionic species, which was readily trapped by Bpin(Oi-Pr) to give boronate 135. A sequential cross-coupling of 135 with two different aryl iodides afforded triarylisoxazole 70 as well.
Fully arylated polyazoles
C–H arylation
Substituted 1,2,3-triazoles are conventionally constructed by using the Huisgen cyclization of alkynes and azides under Cu catalysis.42 The synthesis of multiply arylated 1,2,3-triazoles can be achieved using the same method (Scheme 24). For example, arylacetylenes 136 were reacted with organic azides 137 in the presence of a Cu catalyst to provide 4-aryl-1,2,3-triazole 138, which was then subjected to C–H arylation with arylating agents, giving 4,5-diaryl-1,2,3-triazoles 139. As a pioneering work in the field of C–H arylation of 1,2,3-triazoles, Gevorgyan and coworkers reported a direct Pd-catalyzed arylation of 1,2,3-triazoles with aryl bromides.42a In Pd-catalyzed C–H arylations of 1,2,3-triazoles, aryl chlorides can be used as an arylating reagent under microwave irradiation, which was disclosed by Yorimitsu/Oshima and coworkers.42b Furthermore, Ackermann and coworkers developed Pd-catalyzed C–H arylations of 1,2,3-triazoles with aryl chlorides42c and aryl tosylates.42d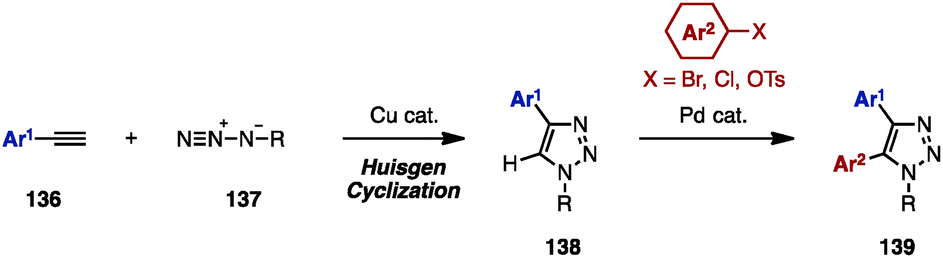 | ||
| Scheme 24 Synthesis of 4,5-diaryl-1,2,3-triazoles by Huisgen cyclization/C–H arylation. | ||
While realizing a SEM-switch strategy (see Schemes 19 and 21), the Sames group also achieved the synthesis of regioisomers of diaryl-1,2,4-triazoles such as (NH)-free diaryltriazole 144 and 4-alkylated diaryltriazole 146 (Scheme 25).43 First, C5-arylation of 1-alkyl-1,2,4-triazoles 140 was accomplished. Treatment of 140 with aryl bromides in the presence of catalytic Pd(OAc)2 and P(n-Bu)Ad2 using a combination of PivOH and K2CO3 (for 140a) or NaOt-Bu (for 140b) provided 5-aryl-1,2,4-triazoles 141a and 141b in high yields. To switch the reactivity for C–H arylation to the C3 position of the 1,2,4-triazole scaffold, SEM or THP groups were transferred from the N1 to the N2 position, upon which a second arylation provided 3,5-diaryl-1,2,4-triazoles 143. The removal of SEM or THP groups under acidic conditions furnished (NH)-free diaryltriazole 144.
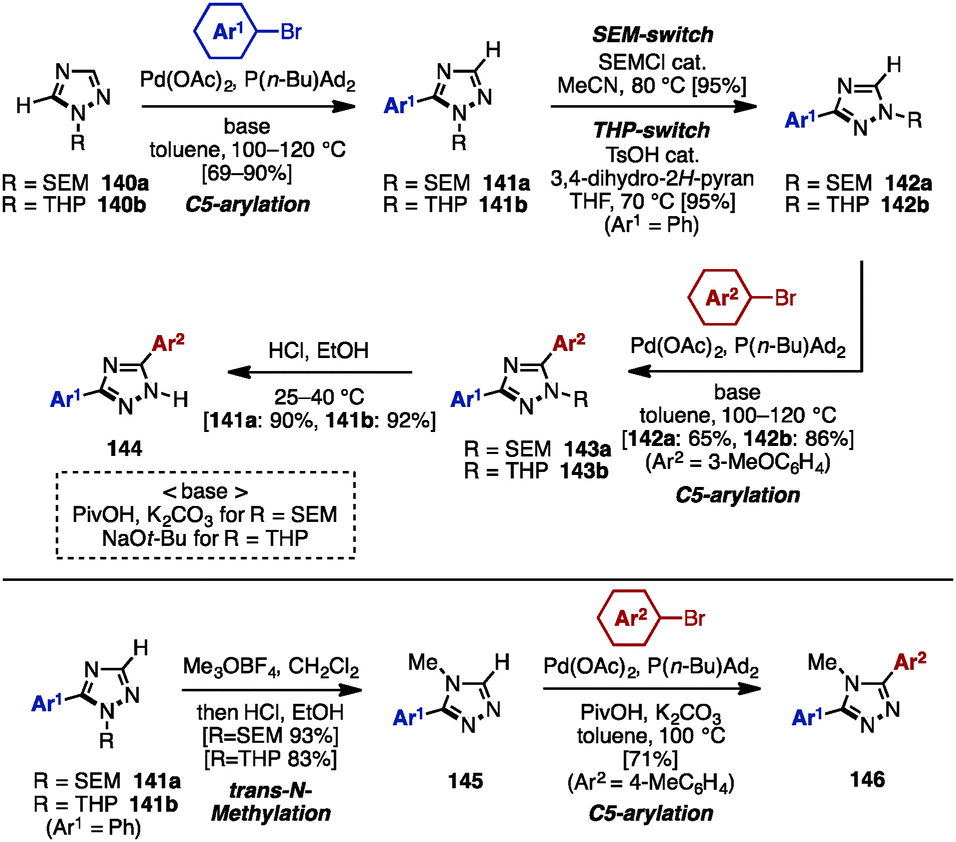 | ||
| Scheme 25 C–H arylation of 1,2,4-triazole by SEM and a THP switch strategy. | ||
In addition, when 1-alkyl-5-aryltriazoles 141 were reacted with Meerwein's reagent (Me3OBF4), trans-N-methylation occurred on the N4 nitrogen atom, giving 4-methyl-4H-triazole 145 as a single regioisomer. Finally, 4-alkylated diaryltriazole 146 was synthesized by one last arylation.
In 2012, Eycken and coworkers reported a Pd/Cu-catalyzed C–H arylation of 1,3,4-thiadiazoles with aryl iodides, bromides, and triflates (Scheme 26A).44 For example, 2-aryl-1,3,4-thiadiazoles 147 was treated with aryl halides in the presence of Pd(OAc)2, Xantphos, CuI, and Cs2CO3 as a base in dioxane to provide 2,5-diaryl-1,3,4-thiadiazoles 148.
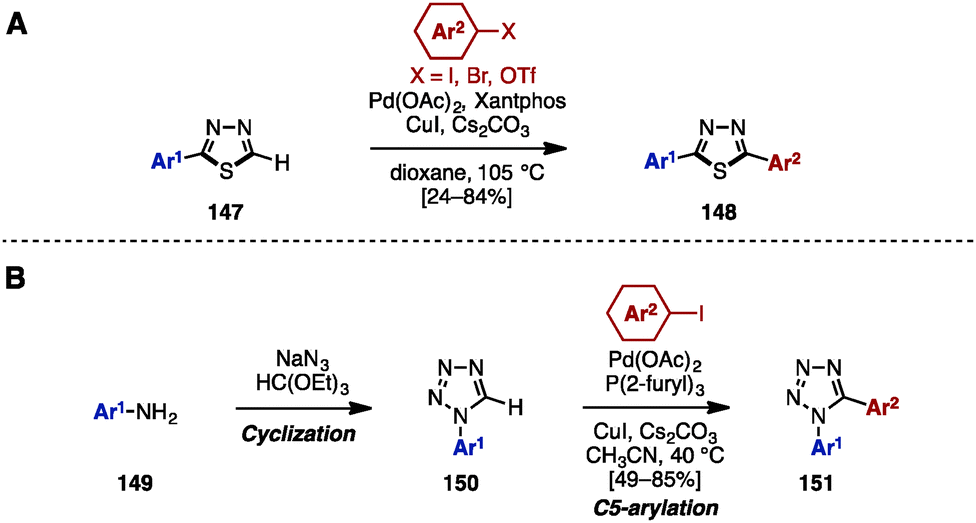 | ||
| Scheme 26 (A) C–H arylation of thiadiazoles by C–H arylation and (B) cycloaddition and C–H arylation for the synthesis of diaryltetrazoles. | ||
In 2010, Pour and coworkers reported a Pd-catalyzed C–H arylation of 1-aryltetrazoles 150 for the synthesis of 1,5-diaryltetrazoles 151 (Scheme 26B).45 By using a cyclization reaction of aniline derivatives 149, sodium azide, and triethyl orthoformate, 1-aryltetrazole 150 was easily prepared. The subsequent C5-arylation of 150 with aryl iodides proceeded in the presence of Pd(OAc)2, P(2-furyl)3, CuI, and Cs2CO3 to provide 1,5-diaryltetrazoles 151. In this catalytic system, the presence of the phosphine ligand is essential to stabilize the intermediate PdII species.
Hexaarylbenzenes (HABs)
Cyclization
Hexaarylbenzene (HAB), a fully arylated form of benzene, is a propeller-shaped, radially π-extended molecule. HABs show exceptional structural diversity when different aryl groups are appended, resulting in a wide range of applications in materials science.46 To synthesize HABs 152 with the same or a few different aryl groups, there are two well-known reactions, involving (1) a [4+2] cycloaddition reaction of tetraarylcyclopentadienones 153 and diarylacetylenes 74 or (2) a metal-catalyzed [2+2+2] cyclotrimerization of 74 (Scheme 27A). For example, in 2007, Hughes and a coworker established a synthetic protocol for unsymmetrical HAB 152 with a maximum of four different aryl groups by preparing unsymmetrical 153 (Scheme 27B).47 The carbonylative coupling of two different benzyl bromide derivatives 154 using Na2Fe(CO)4 (Collman's reagent: 155) as a carbonylative reagent provided unsymmetrical diarylacetones 156. This was then converted to tetraarylcyclopentadienones 153 with three different aryl groups via Knoevenagel condensation with symmetrical diaryldiketones 48. The Diels–Alder reaction of 153 with symmetrical diarylacetylenes 74 furnished HABs 152 bearing four different aryl substituents. The key to making such highly substituted HABs is the unsymmetrical carbonylative coupling step: since unsymmetrical ketones 156 can only be obtained if the reaction of the first benzyl bromide with Collman's reagent is complete before addition of the second benzyl bromide, careful examination of the reaction rates were necessary.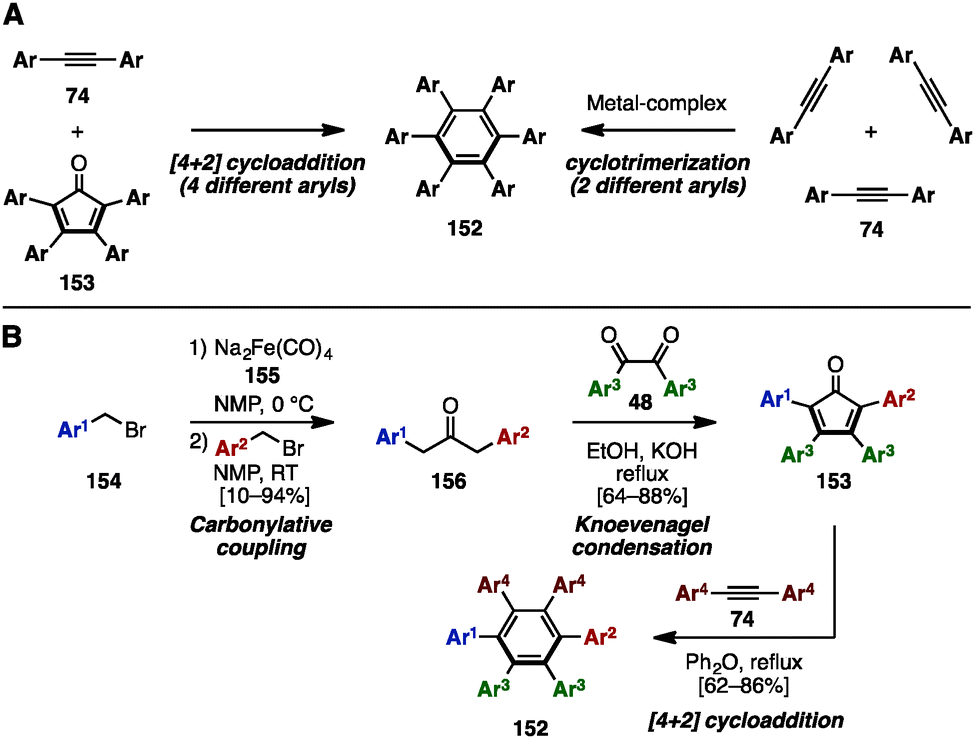 | ||
| Scheme 27 (A) Conventional methods for the synthesis of HABs and (B) a representative protocol for unsymmetrical HABs. | ||
Cross-coupling and C–H arylation
In 2008, Mullen and coworkers reported the synthesis of symmetrically and unsymmetrically substituted HABs 152 by using Hart's benzyne-mediated arylation protocol48 and Suzuki–Miyaura cross-coupling reactions (Scheme 28).49 Treatment of 1,2,4,5-tetrabromo-3,6-dichlorobenzene (157) with excess arylmagnesium bromide led to the formation of a dimagnesium intermediate 158, which was directly quenched with iodine to provide tetraarylated diiodobenzenes 159.49 Subsequent cross-coupling with excess arylboronic acid (4.0 equiv.) using Aliquat 336 as a phase-transfer catalyst in the presence of Pd(PPh3)4 and K2CO3 in toluene at 100 °C provided symmetrically substituted HABs 152 in high yields. In contrast, treatment of 159 with only 1.0 equivalent of arylboronic acid at 80 °C furnished mono-coupled product 160, which was then coupled with another arylboronic acid to afford unsymmetrical HABs 152 bearing four different aryl groups. As HABs are promising precursors for the synthesis of hexabenzocoronenes (HBC), which are privileged structures in materials science, dehydrogenative cyclization of HAB 152a was demonstrated by using FeCl3, producing the corresponding HBC 161.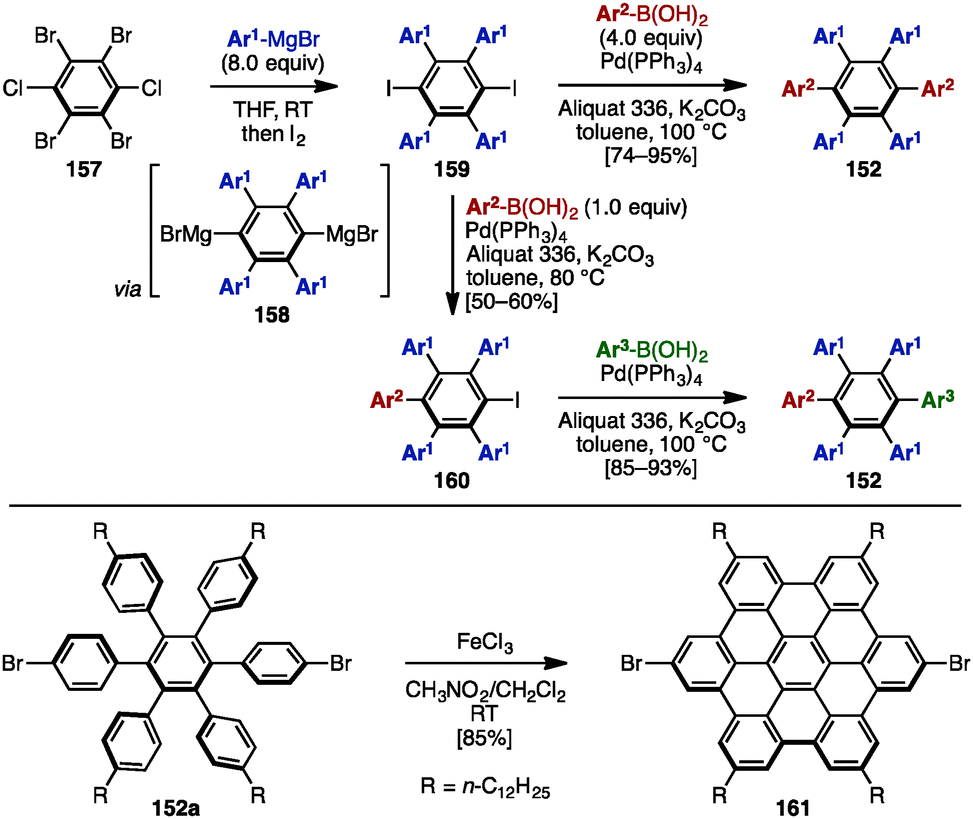 | ||
| Scheme 28 Synthesis of symmetrical and unsymmetrical HABs by using Hart's benzyne-mediated arylation protocol and Suzuki–Miyaura cross-coupling. | ||
In 2015, Yamaguchi, Itami and coworkers achieved the synthesis of HABs with five or six different substituents using C–H arylation, cross-coupling and [4+2] cycloaddition (Scheme 29).50 Initially, their previous synthesis of tetraarylthiophenes 17 with four different aryl groups (see Scheme 9) was modified to allow for a scalable synthesis. First, the Rh-catalyzed C–H arylation of 3-methoxythiophene (43) was changed to a bromination/Suzuki–Miyaura cross-coupling to eliminate the use of an expensive Rh-catalyst. Next, the Pd-catalyzed C–H arylation of 44 with iodoarenes was changed to a Pd-catalyzed C–H arylation of 44 with arylboronic acids in order to achieve better β-selectivity at lower temperatures. After these modifications, a gram-scale synthesis of tetraarylthiophenes 17 was achieved. Then, treatment of 17 with m-chloroperoxybenzoic acid (m-CPBA) in the presence of BF3·OEt2 oxidized the thiophene to the corresponding thiophene S-oxide 162 to enhance the reactivity of the thiophene moiety as a diene. Subsequent [4+2] cycloaddition of 162 with symmetrical diarylacetylenes 74 at 160 °C provided HABs 152b with five different aryl groups. When unsymmetrical diarylacetylene 74 was employed, HABs 152c with six different aryl substituents were synthesized as a mixture of regioisomers. Regioisomers can be separated by chromatography, and the structure of 152c was assigned by X-ray crystal structure analysis. This was the first example of a synthesis of HAB with five or six different aryl groups in a programmable manner.
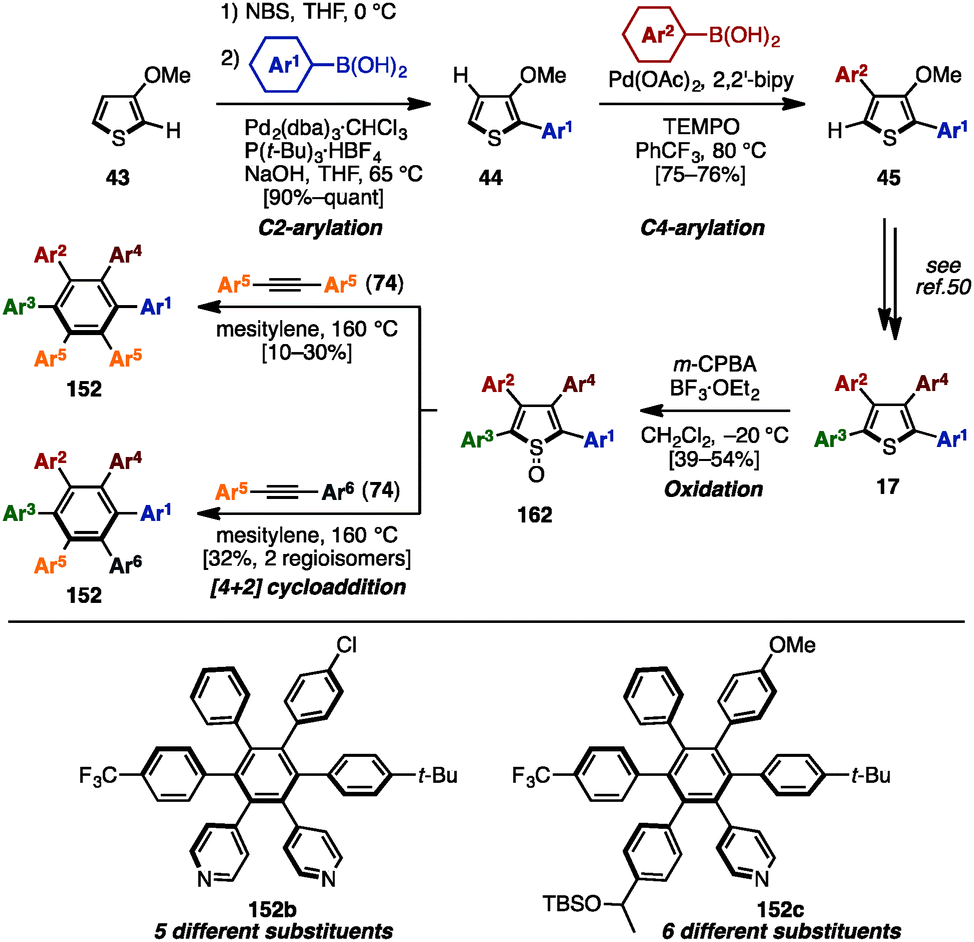 | ||
| Scheme 29 Synthesis of HABs through [4+2] cycloaddition of tetraarylthiophene S-oxides with diarylacetylenes. | ||
In 2016, Jux and coworkers presented a multi-gram synthesis of uncommon HABs starting from p-nitroaniline (163) by utilizing a combination of electrophilic halogenation, Sandmeyer bromination, and Suzuki–Miyaura cross-coupling reactions, calling it a “functionalization of para-nitroaniline” (FpNA) (Scheme 30).51 This synthetic protocol enabled the preparation of 26 different substitution patterns of HABs, of which 18 geometries including five different aryl groups were inaccessible by means of well-established methods. Moreover, this strategy was applicable to the large-scale synthesis of HABs (e.g., 152d was synthesized on an 8.0 g scale).
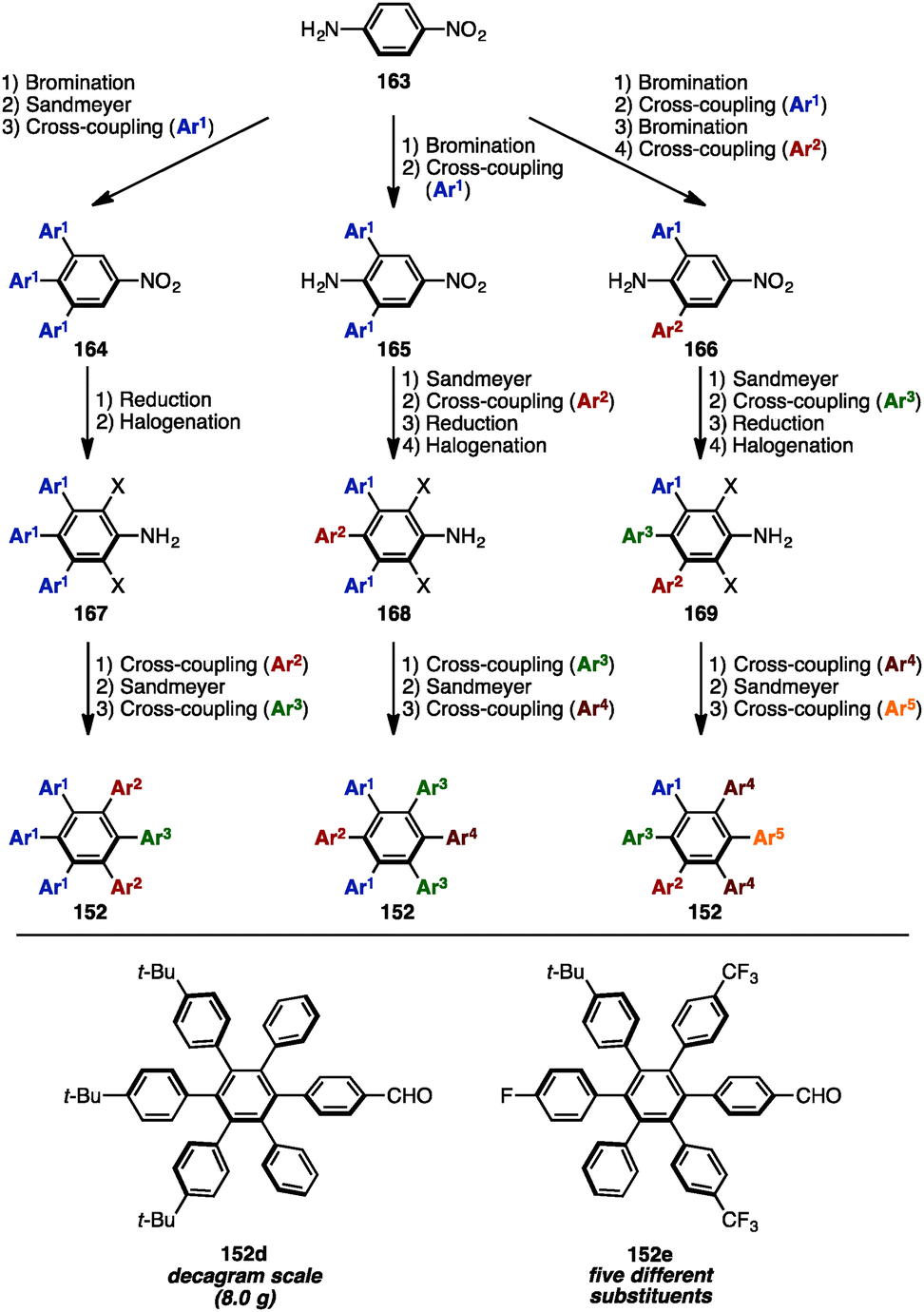 | ||
| Scheme 30 Synthesis of uncommon HABs by a “FpNA protocol”. | ||
Fully arylated pyridines and pyrimidines
Cyclization
Adding to its utility in benzene synthesis, the [4+2] cycloaddition reaction is a powerful tool to construct pyridines. In 2001, Palacios and coworkers reported the dimerization of 2-azadienes 170 in the presence of lithium perchlorate or under thermal conditions for the synthesis of pentaarylpyridines 172 (Scheme 31A).52a In this reaction, 2-azadienes 170 worked as both a dienophile and a diene to dimerize into tetrahydropyridines 171via [4+2] cycloaddition, which was readily converted to the corresponding pyridines 172 or dihydropyridines accompanied by an elimination of imines and/or oxidation. Under lithium perchlorate conditions, dihydropyridines could be obtained (depending on the substituents on the pyridine core), which were immediately oxidized with benzoquinone to give pentaarylpyridines 172. Additionally, in 2008, Draper and coworkers synthesized pentaarylpyridine 172 using a [4+2] cycloaddition reaction of tetraarylcyclopentadienone 153 (tetracyclone: Ar1 = Ph) with arylnitrile (Ar2 = 2-pyridyl) 173 as a dienophile (Scheme 31B).52b To this end, treatment of 153 with 2-arylnitrile 173 under reflux conditions provided pentaarylpyridine 172 with two different aryl groups in excellent yield. Furthermore, in 2015, Yamaguchi and Itami demonstrated a pentaarylpyridine synthesis by [4+2] cyclization of thiophene S-oxides 162 (see Scheme 29) with arylnitriles 173 (Scheme 31C). Although the yields and regioselectivities for the [4+2] cycloaddition were low, this protocol allowed for the synthesis of pentaarylpyridines 172 such as 172a with five different aryl groups.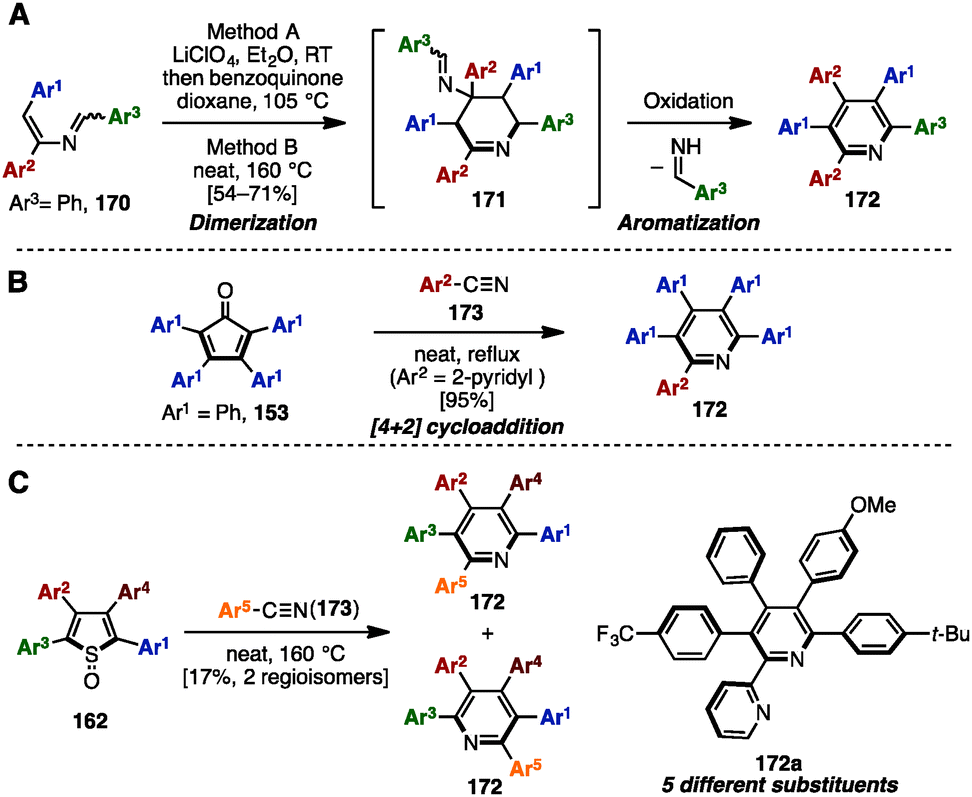 | ||
| Scheme 31 Synthesis of pentaarylpyridines by [4+2] cyclization reactions. | ||
In 2009, Tu and coworkers reported a microwave-assisted multicomponent reaction for pentaarylpyridines 172 from 1,2-diphenylethanone (13) and benzaldehyde derivatives 65 (Scheme 32).53 Treatment of 13 with several benzaldehyde derivatives 64 in the presence of NH4OAc and H2O under microwave irradiation at 170 °C furnished pentaarylpyridines 172 in moderate yields.
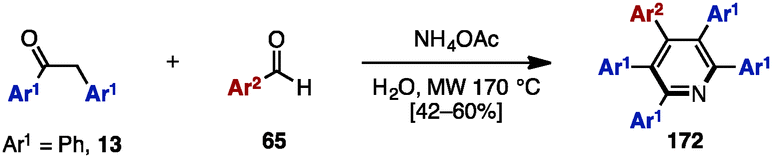 | ||
| Scheme 32 Microwave-assisted pyridine synthesis. | ||
In the case of fully arylated pyrimidines, in 2005, Kiselyov reported a one-pot synthesis of polysubstituted pyrimidines including tetraarylated pyrimidine 178a by cyclization of in situ-generated α,β-unsaturated imines and amidines (Scheme 33).54 This one-pot sequence began with the nucleophilic addition of phosphorus ylide 174 to arylnitrile 173a to yield ylide 175. Subsequent Horner–Wadsworth–Emmons reaction with aldehyde 65a furnished α,β-unsaturated imine 176. Lastly, 176 was cyclized with amidine 177 in the presence of i-Pr2NEt to provide tetraarylpyrimidine 178a with four different aryl groups, albeit in low yield.
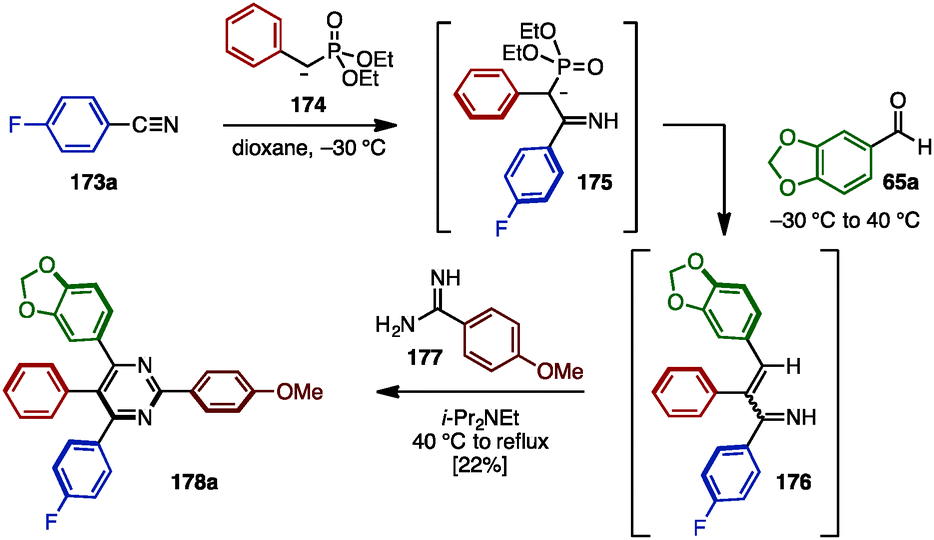 | ||
| Scheme 33 Cyclization of in situ-generated α,β-unsaturated imine and amidine. | ||
In 2013, Liu and coworkers reported zirconium-mediated multicomponent reactions of diarylacetylene 74 with arylnitriles 173, which enabled a regioselective synthesis of tetraarylpyrimidines 178 (Scheme 33).55 Diarylacetylenes 74 reacted with zirconocene 179 to form a phosphine-stabilized zirconocene–alkyne complex 180, which was readily coupled with aryl nitriles 173, giving tetraarylpyrimidines 178 in moderate yields (Scheme 34).
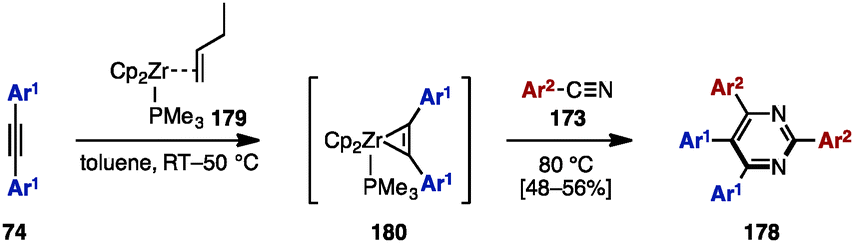 | ||
| Scheme 34 Zirconium-mediated multicomponent reactions. | ||
Cross-coupling
Similarly to the synthesis of tetraarylpyrroles and thiophenes (see Scheme 4), in 2014, Langer and coworkers synthesized a series of pentaarylpyridines by multiple Suzuki–Miyaura cross-coupling reactions of pentachloropyridine (181) (Scheme 35A).56 Treatment of 181 with excess arylboronic acid in the presence of PdCl2(MeCN)2, S-Phos, and K3PO4 in toluene at 100 °C (method A) provided pentaaarylpyridines 172 with one type of aryl group. When the reaction was performed with fewer equivalents of arylboronic acid under Pd(PPh3)4 and K3PO4 in MeCN at 80 °C (method B), the cross-coupling reaction occurred at only the C2- and C6-positions of the pyridine scaffold. Then, by switching the solvent from MeCN to toluene, and increasing the reaction temperature (method C), pentaarylpyridines 172 with two different aryl substituents were successfully synthesized. Moreover, starting from 4-aryl-2,3,5,6-tetrachloropyridine 183, this sequential cross-coupling protocol provided pentaarylpyridines 172 with two or three different aryl groups in one- or two-step operations. In addition, Langer and coworkers reported the synthesis of tetraarylpyrimidines 178 from tetrachloropyrimidine (186) in a similar manner (Scheme 35B).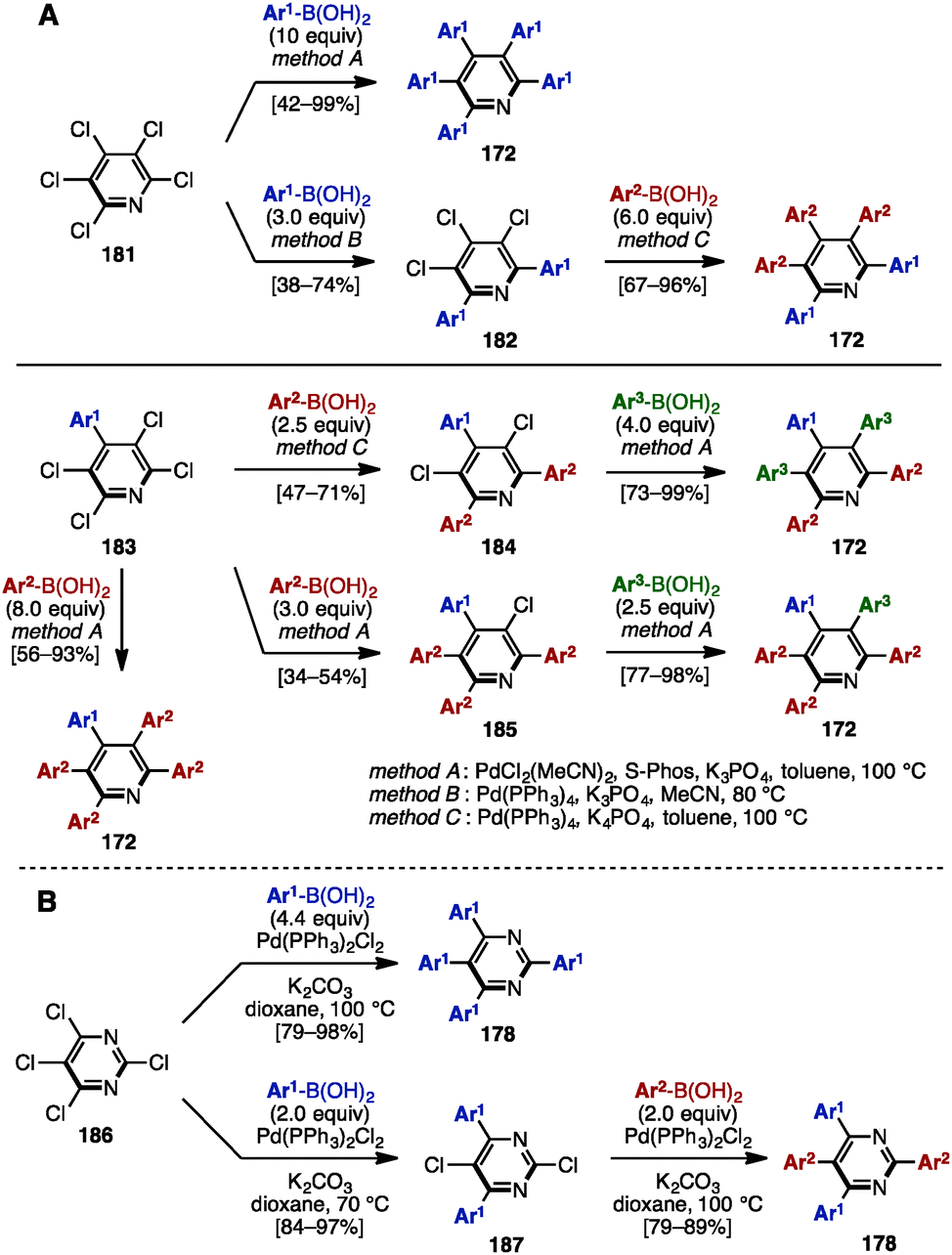 | ||
| Scheme 35 (A) Synthesis of pentaarylpyridines by multiple Suzuki–Miyaura cross-couplings and (B) synthesis of tetraarylpyrimidines in a similar manner. | ||
In 2014, Schmitt and coworkers established a synthetic route toward multiply arylated pyridines bearing up to five different aryl groups, which involved five-fold sequential and regioselective Suzuki–Miyaura cross-coupling reactions starting from commercially available 2-chloro-3-hydroxypyridine (188) (Scheme 36).57 2,4,6-Trihalogenated pyridine 189, which was readily prepared from 188 in two steps, was coupled with two different arylboronic acids to furnish 4,6-diarylpyridine 190 regioselectively. Then, installation of a benzyloxy group, removal of a MOM group, and triflation of the resulting hydroxy group afforded 2-(benzyloxy)pyridine 191. The third cross-coupling event converted 191 into 3,4,6-triarylpyridine 192, which was then brominated and cross-coupled at the C5-position to provide 3,4,5,6-tetraarylpyridine 193. Finally, after removal of the benzyl group, the resulting 2-hydroxypyridine was triflated and coupled with an arylboronic acid to yield pentaarylpyridines 172 such as 172a with five different substituents in a total of 13 steps.
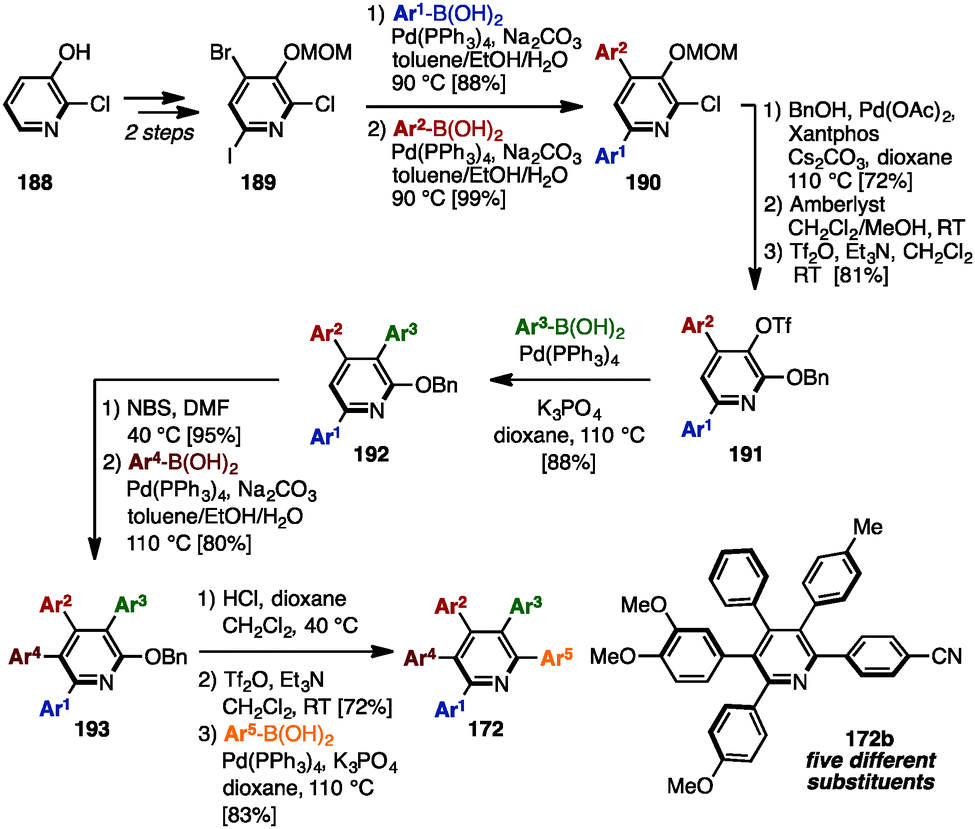 | ||
| Scheme 36 Synthesis of pentaarylpyridines with five different aryl groups by fully regiocontrolled Suzuki–Miyaura cross-coupling. | ||
Others
In 2000, Pascal and coworkers synthesized octaarylnaphthalene 195 with two different aryl substituents by [4+2] cycloaddition of tetraarylcyclopentadienone 153 and tetraarylbenzyne (Scheme 37).58 The [4+2] cycloaddition of 153 (Ar1 = Ph, Ar2 = 4-tolyl) with tetraarylbenzyne, which was readily generated by treatment of tetraarylanthranilic acid 194 with isoamyl nitrile, provided octaarylnaphthalene 195 with two different aryl groups in 83% yield.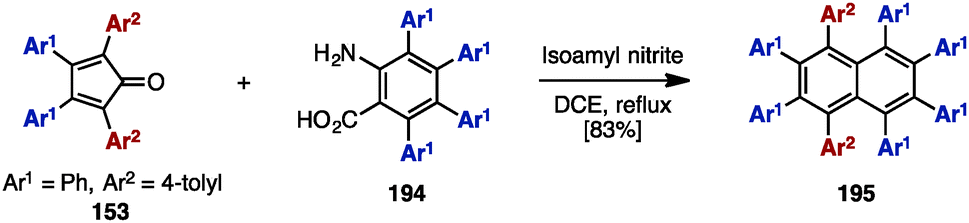 | ||
| Scheme 37 [4+2] cycloaddition of tetraarylcyclopentadienone and tetraarylbenzyne. | ||
In the 1990s, syntheses of heptaarylisoindole 19759a and heptaarylisoquinoline 19859b were reported by Hay and coworkers via the condensation of 1,2-bis(benzoyl)benzene 196 with aniline derivatives 146 or benzylamine derivative 48, respectively (Scheme 38). Treatment of 196 (Ar1 = Ph, Ar2 = 4-PhOC6H4) with excess 146 (Ar3 = 4-MeC6H4) using p-toluenesulfonic acid (p-TsOH) at 200 °C furnished isoindole 197 in 90% yield. When benzylamine 49 was reacted with 196 and DBU in chlorobenzene under reflux conditions, heptaarylated isoquinoline 198 was also synthesized (41% yield).
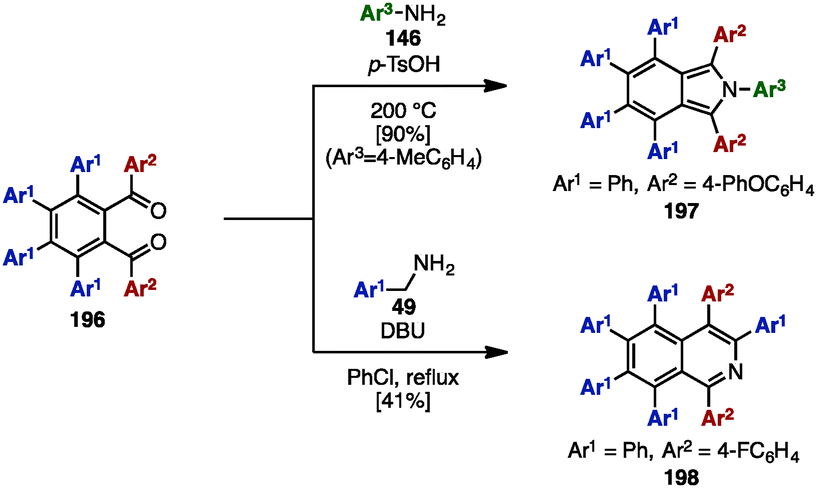 | ||
| Scheme 38 Synthesis of hexaarylisoindole and heptaarylisoquinoline. | ||
In 2009, Miura and coworkers reported Pd-catalyzed oxidative coupling reactions of N-substituted pyrroles and their carboxylic acid derivatives with diarylacetylenes (Scheme 39).60 Treatment of 199 with diarylacetylene 74 in the presence of Pd(OAc)2, Cu(OAc)2, LiOAc, and MS4Å in dimethylacetamide (DMA) provided tetraarylated indole 200. When using Ag2CO3 and 2,6-dimethylbenzoic acid as an oxidant and an additive instead of Cu(OAc)2·H2O and LiOAc, the second coupling reaction of 200 with diarylacetylenes 74 proceeded to furnish octaarylcarbazoles 201 with two different aryl substituents.
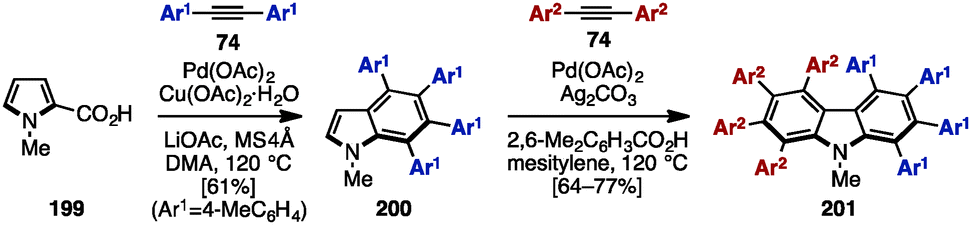 | ||
| Scheme 39 Synthesis of multiply arylated carbazoles using Pd-catalyzed oxidative coupling reactions with diarylalkynes. | ||
Conclusions
This article summarized methodologies developed for the synthesis of fully arylated arenes with more than two different aryl substituents, including 5-membered (hetero)arenes, 6-membered (hetero)arenes, and fused polycyclic (hetero)arenes. Although the synthesis of structurally beautiful but complex fully arylated arenes has been facilitated over many decades, application of these molecules in materials and biological sciences is still rare. It is our hope that new methodologies to access unexploited molecules will continue to aid the discovery of new functional materials.Acknowledgements
This work was supported by JSPS KAKENHI Grant No. JP16H01011, JP16H04148, JP16K13085, and 16H01140 (to J. Y.), and a JSPS research fellowship for young scientists (to S. S.). We thank Dr Yoshihiro Ishihara (Vertex Pharmaceuticals) for fruitful discussion and critical comments.Notes and references
- Selected examples, (a) E. L. Williams, K. Haavisto, J. Li and G. E. Jabbour, Adv. Mater., 2007, 19, 197 CrossRef CAS; (b) F. Li, Z. Chen, W. Wei, H. Cao, Q. Gong, F. Teng, L. Qian and Y. Wang, J. Phys. D: Appl. Phys., 2004, 37, 1613 CrossRef CAS; (c) B. S. Ong, Y. Wu, P. Liu and S. Gardner, J. Am. Chem. Soc., 2004, 126, 3378 CrossRef CAS PubMed; (d) K. Shin-ya, K. Wierzba, K. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Sato, J. Am. Chem. Soc., 2001, 123, 1262 CrossRef CAS PubMed.
- R. Rossi, F. Bellina, M. Lessi, C. Manzini and L. Perego, Synthesis, 2014, 2833 CrossRef.
- Selected examples, (a) K. R. J. Thomas, M. Velusamy, J. T. Lin, C. H. Chuen and Y. T. Tao, J. Mater. Chem., 2005, 15, 4453 RSC; (b) J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys. Lett., 1993, 63, 2627 CrossRef CAS; (c) K. Suzuki K. Ueno A. Senoo JP. Pat.,109765 A, 2003 Search PubMed; (d) S. Miwatashi, Y. Arikawa, K. Naruo, K. Igaki, Y. Watanabe, H. Kimura, T. Kawamoto and S. Ohkawa, Chem. Pharm. Bull., 2005, 53, 410 CrossRef CAS PubMed; (e) N. W. Johnson, M. Semones, J. L Adams, M. Hansbury and J. Winkler, Bioorg. Med. Chem. Lett., 2007, 17, 5514 CrossRef CAS PubMed; (f) S. H. Lee, H. J. Seo, M. J. Kim, S. Y. Kang, S.-H. Lee, K. Ahn, M. Lee, H.-K. Han, J. Kim and J. Lee, Bioorg. Med. Chem. Lett., 2009, 19, 6632 CrossRef CAS PubMed.
- (a) L. Knorr, Ber., 1884, 17, 2863 CrossRef; (b) C. Paal, Ber., 1884, 17, 2756 CrossRef.
- (a) W.-J. Kuo, Y.-H. Chen, R.-J. Jeng, L.-H. Chan, W.-P. Lin and Z.-M. Yang, Tetrahedron, 2007, 63, 7086 CrossRef CAS; (b) C.-S. Li, Y.-H. Tsai, W.-C. Lee and W.-J. Kuo, J. Org. Chem., 2010, 75, 4004 CrossRef CAS PubMed; (c) S. Mao, X.-Q. Zhu, Y.-R. Gao, D.-D. Guo and Y.-Q. Wang, Chem. – Eur. J., 2015, 21, 11335 CrossRef CAS PubMed; (d) S. Mao, Y.-R. Gao, S.-L. Zhang, D.-D. Guo and Y.-Q. Wang, Eur. J. Org. Chem., 2015, 876 CrossRef CAS.
- I. Bergner and T. Opatz, J. Org. Chem., 2007, 72, 7083 CrossRef CAS PubMed.
- X. Wu, K. Li, S. Wang, C. Liu and A. Lei, Org. Lett., 2016, 18, 56 CrossRef CAS PubMed.
- (a) T. T. Dang, R. Ahmad, T. T. Dang, H. Reinke and P. Langer, Tetrahedron Lett., 2008, 49, 1698 CrossRef CAS; (b) F. Yang, T. Jin, M. Bao and Y. Yamamoto, Tetrahedron, 2011, 67, 10147 CrossRef CAS; (c) Y. Dang and Y. Chen, J. Org. Chem., 2007, 72, 6901 CrossRef CAS PubMed; (d) T. T. Dang, N. Rasool, T. T. Dang, H. Reinke and P. Langer, Tetrahedron Lett., 2007, 48, 845 CrossRef CAS; (e) Đ. T. Tùng, Đ. T. Tuân, N. Rasool, A. Villinger, H. Reinke, C. Fischer and P. Langer, Adv. Synth. Catal., 2009, 351, 1595 CrossRef.
- A. Rahimi, J. C. Namyslo, M. H. H. Drafz, J. Halm, E. Hübner, M. Nieger, N. Rautzenberg and A. Schmidt, J. Org. Chem., 2011, 76, 7316 CrossRef CAS PubMed.
- F. Yang, T. Jin, M. Bao and Y. Yamamoto, Tetrahedron, 2011, 67, 10147 CrossRef CAS.
- (a) S. Ishikawa, Y. Noda, M. Wada and T. Nishikata, J. Org. Chem., 2015, 80, 7555 CrossRef CAS PubMed; (b) T. Nishikata, Y. Yamane, Y. Yamaguchi and S. Ishikawa, Asian J. Org. Chem., 2016, 5, 466 CrossRef CAS.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) L.-C. Campeau, R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35 CAS; (c) T. Satoh and M. Miura, Chem. Lett., 2007, 36, 200 CrossRef CAS; (d) I.-V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (e) I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036 RSC; (f) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (g) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269 CrossRef CAS; (h) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (i) O. Daugulis, Top. Curr. Chem., 2010, 292, 57 CrossRef CAS PubMed; (j) J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20 CrossRef CAS; (k) M. Livendahl and A. M. Echavarren, Isr. J. Chem., 2010, 50, 630 CrossRef CAS; (l) Y.-X. Su and L.-P. Sun, Mini-Rev. Org. Chem., 2012, 9, 87 CrossRef CAS; (m) L. Ackermann, A. R. Kapdi, H. K. Potuchi and S. I. Kozhushkov, Handbook of Green Chemistry, Wiley-VCH, Weinheim, 2012, vol. 7, p. 259 Search PubMed; (n) A. Sharma, D. Vacchani and E. Van der Eycken, Chem. – Eur. J., 2013, 19, 1158 CrossRef CAS PubMed; (o) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17 CrossRef CAS.
- M. Nakano, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1851 CrossRef CAS PubMed.
- S. Yanagisawa, K. Ueda, H. Sekizawa and K. Itami, J. Am. Chem. Soc., 2009, 131, 14622 CrossRef CAS PubMed.
- (a) S. Yanagisawa, T. Sudo, R. Noyori and K. Itami, J. Am. Chem. Soc., 2006, 128, 11748 CrossRef CAS PubMed; (b) K. Ueda, K. Amaike, R. M. Maceiczyk, K. Itami and J. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 13226 CrossRef CAS PubMed; (c) A. D. Yamaguchi, K. M. Chepiga, J. Yamaguchi, K. Itami and H. M. L. Davies, J. Am. Chem. Soc., 2015, 137, 644 CAS.
- S. Yanagisawa and K. Itami, Tetrahedron, 2011, 67, 4425 CrossRef CAS.
- (a) S. Samanta, D. Roy, S. Khamarui and D. K. Maiti, Chem. Commun., 2014, 50, 2477 RSC; (b) R. Sarkar and C. Mukhopadhyay, Eur. J. Org. Chem., 2015, 1246 CrossRef CAS; (c) R. Sarkar and C. Mukhopadhyay, Tetrahedron Lett., 2015, 56, 3872 CrossRef CAS; (d) P. Hu, Q. Wang, Y. Yan, S. Zhang, B. Zhang and Z. Wang, Org. Biomol. Chem., 2013, 11, 4304 RSC.
- (a) W. Pei, S. Li, X. Nie, Y. Li, J. Pei, B. Chen, J. Wu and X. Ye, Synthesis, 1998, 1298 CrossRef CAS; (b) P. C. Patil, F. A. Luzzio and D. R. Demuth, Tetrahedron Lett., 2015, 56, 3039 CrossRef CAS PubMed.
- C. Kison and T. Opatz, Chem. – Eur. J., 2008, 15, 843 CrossRef PubMed.
- (a) H. Bredereck, R. Gompper and D. Hayer, Chem. Ber., 1953, 86, 88 CrossRef CAS; (b) H. Bredereck and R. Bangert, Angew. Chem., Int. Ed. Engl., 1962, 1, 662 CrossRef.
- (a) J. L. Bailey and R. R. Sudini, Tetrahedron Lett., 2014, 55, 3674 CrossRef CAS; (b) J. S. Carter, D. J. Rogier, M. J. Graneto, K. Seibert, C. M. Koboldt, Y. Zhang and J. J. Talley, Bioorg. Med. Chem. Lett., 1999, 9, 1167 CrossRef CAS PubMed.
- Y.-L. Tsai, Y.-S. Fan, C.-J. Lee, C.-H. Huang, U. Das and W. Lin, Chem. Commun., 2013, 49, 10266 RSC.
- J. Xuan, L.-Q. Lu, J. R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2013, 6755 CrossRef CAS.
- (a) T.-T. Zeng, J. Xuan, W. Ding, K. Wang, L.-Q. Lu and W.-J. Xiao, Org. Lett., 2015, 17, 4070 CrossRef CAS PubMed; (b) T. Chatterjee, J. Y. Cho and E. J. Cho, J. Org. Chem., 2016, 81, 6995 CrossRef CAS PubMed.
- F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583 CrossRef CAS.
- (a) R. Shetty, S. Shafi and Y. Kuberan, J. Pharma Res., 2013, 6, 897 CrossRef CAS; (b) S. E. Denmark and J. M. Kallemeyn, J. Org. Chem., 2005, 70, 2839 CrossRef CAS PubMed; (c) S. Kankala, R. Vadde and C. S. Vasam, Org. Biomol. Chem., 2011, 9, 7869 RSC.
- (a) B. Clapham and A. J. Sutherland, J. Org. Chem., 2001, 66, 9033 CrossRef CAS PubMed; (b) K. J. Hodgetts and M. T. Kershaw, Org. Lett., 2002, 4, 2905 CrossRef CAS PubMed; (c) I. C. Christoforou and P. A. Koutentis, Org. Biomol. Chem., 2007, 5, 1381 RSC.
- C. Sämann, E. Coya and P. Knochel, Angew. Chem., Int. Ed., 2013, 53, 1430 CrossRef PubMed.
- D. Haas, M. Mosrin and P. Knochel, Org. Lett., 2013, 15, 6162 CrossRef CAS PubMed.
- M. McLaughlin, K. Marcantonio, C.-Y. Chen and I. W. Davies, J. Org. Chem., 2008, 73, 4309 CrossRef CAS PubMed.
- (a) L.-C. Campeau, M. Bertrand-Laperle, J.-P. Leclerc, E. Villemure, S. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3276 CrossRef CAS PubMed; (b) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef CAS PubMed.
- J. M. Joo, B. B. Touré and D. Sames, J. Org. Chem., 2010, 75, 4911 CrossRef CAS PubMed.
- (a) F. Shibahara, E. Yamaguchi and T. Murai, J. Org. Chem., 2011, 76, 2680 CrossRef CAS PubMed; (b) F. Shibahara, T. Yamauchi, E. Yamaguchi and T. Murai, J. Org. Chem., 2012, 77, 8815 CrossRef CAS PubMed.
- A. Yokooji, T. Okazawa, T. Satoh, M. Miura and M. Nomura, Tetrahedron, 2003, 59, 5685 CrossRef CAS.
- S. Tani, T. N. Uehara, J. Yamaguchi and K. Itami, Chem. Sci., 2014, 5, 123 RSC.
- (a) G. L. Turner, J. A. Morris and M. F. Greaney, Angew. Chem., Int. Ed., 2007, 46, 7996 CrossRef CAS PubMed; (b) S. K. Kim, J.-H. Kim, Y. C. Park, J. W. Kim and E. K. Yum, Tetrahedron, 2013, 69, 10990 CrossRef CAS.
- R. Goikhman, T. L. Jacques and D. Sames, J. Am. Chem. Soc., 2009, 131, 3042 CrossRef CAS PubMed.
- T. Morita, D. Kobayashi, K. Matsumura, K. Johmoto, H. Uekusa, S. Fuse and T. Takahashi, Chem. – Asian J., 2015, 10, 1626 CrossRef CAS PubMed.
- S. Fuse, T. Morita, K. Johmoto, H. Uekusa and H. Tanaka, Chem. – Eur. J., 2015, 21, 14370 CrossRef CAS PubMed.
- T. Morita, S. Fuse and H. Nakamura, Angew. Chem., Int. Ed., 2016, 55, 13580 CrossRef CAS PubMed.
- M. Shigenobu, K. Takenaka and H. Sasai, Angew. Chem., Int. Ed., 2015, 54, 9572 CrossRef CAS PubMed.
- (a) S. Chuprakov, N. Chernyak, A. S. Dudnik and V. Gevorgyan, Org. Lett., 2007, 9, 2333 CrossRef CAS PubMed; (b) M. Iwasaki, H. Yorimitsu and K. Oshima, Chem. – Asian J., 2007, 2, 1430 CrossRef CAS PubMed; (c) L. Ackermann, R. Vicente and R. Born, Adv. Synth. Catal., 2008, 350, 741 CrossRef CAS; (d) L. Ackermann, A. Althammer and S. Fenner, Angew. Chem., Int. Ed., 2008, 48, 201 CrossRef PubMed.
- J. M. Joo, P. Guo and D. Sames, J. Org. Chem., 2013, 78, 738 CrossRef CAS PubMed.
- D. D. Vachhani, A. Sharma and E. Van der Eycken, J. Org. Chem., 2012, 77, 8768 CrossRef CAS PubMed.
- M. Špulák, R. Lubojacký, P. Šenel, J. Kuneš and M. Pour, J. Org. Chem., 2010, 75, 241 CrossRef PubMed.
- V. Vij, V. Bhalla and M. Kumar, Chem. Rev., 2016, 116, 9565 CrossRef CAS PubMed.
- R. G. Potter and T. S. Hughes, Org. Lett., 2007, 9, 1187 CrossRef CAS PubMed.
- (a) H. Hart and P. Rajakumar, Tetrahedron, 1995, 51, 1313 CrossRef CAS; (b) K. Harada, H. Hart and C.-J. F. Du, J. Org. Chem., 1985, 50, 5524 CrossRef CAS; (c) P. Rajakumar and A. Kannan, Tetrahedron Lett., 1993, 34, 8317 CrossRef CAS.
- X. Yang, X. Dou and K. Müllen, Chem. – Asian J., 2008, 3, 759 CrossRef CAS PubMed.
- S. Suzuki, Y. Segawa, K. Itami and J. Yamaguchi, Nat. Chem., 2015, 7, 227 CrossRef CAS PubMed.
- D. Lungerich, D. Reger, H. Hölzel, R. Riedel, M. M. J. C. Martin, F. Hampel and N. Jux, Angew. Chem., Int. Ed., 2016, 55, 5602 CrossRef CAS PubMed.
- (a) F. Palacios, C. Alonso, G. Rubiales and J. M. Ezpeleta, Eur. J. Org. Chem., 2001, 2115 CrossRef CAS; (b) C. M. A. Ollagnier, S. D. Perera, C. M. Fitchett and S. M. Draper, Dalton Trans., 2008, 283 RSC.
- B. Jiang, W.-J. Hao, X. Wang, F. Shi and S.-J. Tu, J. Comb. Chem., 2009, 11, 846 CrossRef CAS PubMed.
- A. S. Kiselyov, Tetrahedron Lett., 2005, 46, 1663 CrossRef CAS.
- X. You, S. Yu and Y. Liu, Organometallics, 2013, 32, 5273 CrossRef CAS.
- S. Reimann, P. Ehlers, A. Petrosyan, S. Kohse, A. Spannenberg, A. E. Surkus, T. V. Ghochikyan, A. S. Saghyan, S. Lochbrunner, O. Kühn, R. Ludwig and P. Langer, Adv. Synth. Catal., 2014, 356, 1987 CrossRef CAS.
- C. Doebelin, P. Wagner, F. Bihel, N. Humbert, C. A. Kenfack, Y. Mely, J.-J. Bourguignon and M. Schmitt, J. Org. Chem., 2014, 79, 908 CrossRef CAS PubMed.
- R. A. Pascal Jr., L. Barnett, A. X. Qiao and D. M. Ho, J. Org. Chem., 2000, 65, 7711 CrossRef.
- (a) Y. Ding and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3293 CrossRef CAS; (b) R. Singh and A. S. Hay, Macromolecules, 1992, 25, 1033 CrossRef CAS.
- M. Yamashita, H. Horiguchi, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7481 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |