
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-orthogonal “click-and-release” donation of caged carbonyl sulfide (COS) and hydrogen sulfide (H2S)†
Andrea K.
Steiger‡
 a,
Yang
Yang‡
b,
Maksim
Royzen
*b and
Michael D.
Pluth
*a
a,
Yang
Yang‡
b,
Maksim
Royzen
*b and
Michael D.
Pluth
*a
aMaterials Science Institute, Institute of Molecular Biology, Department of Chemistry and Biochemistry, University of Oregon, Eugene, OR 97403, USA. E-mail: pluth@uoregon.edu
bDepartment of Chemistry, University at Albany, SUNY, 1400 Washington Avenue, Albany, NY 12222, USA. E-mail: mroyzen@albany.edu
First published on 22nd December 2016
Abstract
Hydrogen sulfide (H2S) is an important biomolecule with high therapeutic potential. Here we leverage the inverse-electron demand Diels–Alder (IEDDA) click reaction between a thiocarbamate-functionalized trans-cyclooctene and a tetrazine to deliver carbonyl sulfide (COS), which is quickly converted to H2S by the uniquitous enzyme carbonic anhydrase (CA), thus providing a new strategy for bio-orthogonal COS/H2S donation.
With the recent addition of hydrogen sulfide (H2S) to the list of biologically-relevant gasotransmitters,1 significant efforts have focused on developing H2S donors as powerful research, and potentially therapeutic, tools.2,3 Endogenous H2S production occurs primarily from cystathionine-γ-lyase (CSE), cystathionine-β-synthase (CBS), and 3-mercaptopyruvate transferase (3-MST), and the slow production of H2S exerts protective effects throughout the body.1 Although convenient, inorganic sulfide salts (NaSH and Na2S) provide a large, instantaneous bolus of H2S, and sulfide oxidation often occurs rapidly after administration.4 These limitations suggest that more efficacious donors should either more closely mimic slower enzymatic production rates or be stable until triggered to release H2S in response to specific stimuli. Available synthetic slow-release donors have already made major impacts in H2S research, and several small molecule H2S donors have already entered clinical trials.5 Despite this promise, providing temporal control over H2S release remains a major challenge, and there is significant interest in developing synthetic H2S donors that are activated by well-defined triggering mechanisms that enable on-demand H2S release.
Aligned with this need, we recently pioneered the use of carbonyl sulfide (COS)-releasing molecules as a strategy to access responsive H2S donors. We demonstrated that self-immolative thiocarbamates can be triggered to decompose and release COS, which is rapidly converted to H2S by the ubiquitous enzyme carbonic anhydrase (CA).6 Analogous to the broad applications of self-immolative carbamates as delivery platforms for prodrugs, fluorophores, and other biologically-relevant payloads, thiocarbamates provide a highly tunable platform on which the triggering mechanism can be engineered to initiate self-immolation and COS release by specific analytes of interest. Since our initial report on caged COS/H2S release, passive H2S donation from small molecule and polymeric N-thiocarboxyanhydrides7 as well as responsive ROS-triggered donors that provide protection against cellular oxidative stress have been reported.8 Missing from current COS/H2S donor technologies are platforms activated by bio-orthogonal triggers to allow precise temporal control for H2S release. Motivated by this need, we report here the first example of bio-orthogonal activation of COS/H2S release through adaptation of the well-developed inverse-electron demand Diels–Alder (IEDDA) click reaction to release COS/H2S (Scheme 1).
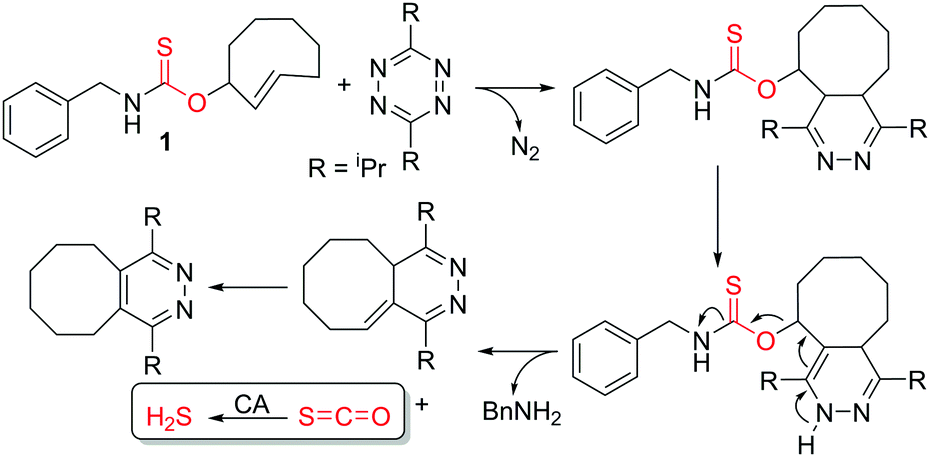 | ||
| Scheme 1 IEDDA reaction of thiocarbamate-functionalized TCO 1 with tetrazine to generate COS/H2S. | ||
The IEDDA reaction between a trans-cyclooctene (TCO) and a tetrazine is a proven platform for bio-orthogonal click reactions in living systems.9–11 In addition to providing an important biocompatible bond-forming tool, the IEDDA reaction has also been adapted for targeted drug release by using functionalized benzylic carbamates, which can be triggered to undergo self-immolative decomposition following the click reaction and subsequently release the attached drug, as well as CO2 as a byproduct.12–14 We envisioned that a similar strategy could be leveraged to develop a fully bio-orthogonal COS/H2S releasing platform by using a benzylic thiocarbamate-functionalized TCO (Scheme 1). The initial IEDDA click reaction would generate the thiocarbamate-functionalized dihydropyridazine, which after tautomerization, deprotonation, and rearomatization can extrude COS, BnNH2, and the cyclooctylpyridazine product. To test this hypothesis, we prepared TCO 1 by treating (E)-cyclooct-2-enol with benzyl isothiocyanate in the presence of NaH.13 In parallel, we prepared the analogous carbamate-functionalized TCO 2, which undergoes the same IEDDA reaction but releases CO2 rather than COS (Scheme 2). Both TCO 1 and 2 are isolated as the axial isomer, which is estimated to be significantly more reactive than the analogous equatorial isomer.15 Importantly, this design strategy provides simple synthetic access to both the thiocarbamate donor and key carbamate control compounds. Additionally, click-and-release CO donors were recently reported utilizing an intramolecular Diels–Alder reaction, thus supporting the validity of our approach for accessing bio-orthogonal gasotransmitter release.16
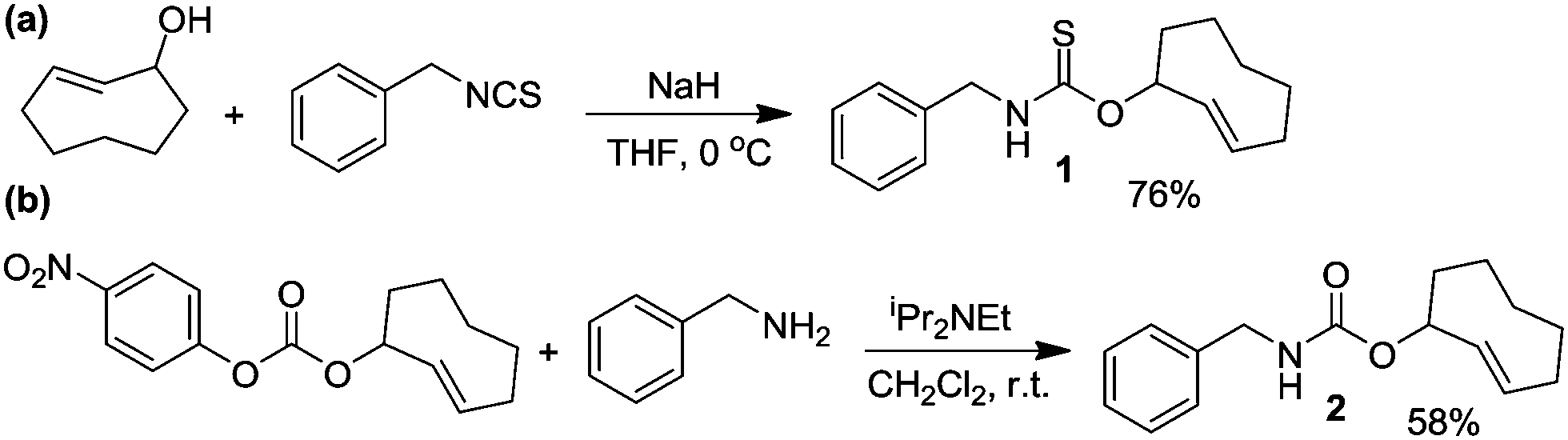 | ||
| Scheme 2 Synthesis of thiocarbamate-functionalized COS/H2S donor TCO 1 and the associated carbamate-functionalized control compound TCO 2. | ||
To confirm that the IEDDA reaction would initiate self-immolative decomposition of the thiocarbamate moiety, we monitored the reaction of 1 and 2 equiv. of bis-isopropyl-1,2,4,5-tetrazine in wet methanol-d4 by 1H NMR spectroscopy (Fig. 1a). Within 5 minutes of tetrazine addition, we observed disappearance of the alkene peaks (5.3–5.7 ppm), indicative of cycloaddition. New resonances corresponding to BnNH2 were subsequently observed at 4.25 ppm, while 1 continues to decompose over 24 hours. Due to the complexity of this reaction and the different potential intermediates that could be formed en route to COS extrusion, we also monitored product formation by mass spectrometry. Consistent with our design hypothesis we observed the re-aromatized IEDDA product (M + H+ 247.239), BnNH2 (M + H+ 108.091), and COS (M + H+ 61.044) (Fig. 1b and c) using direct analysis in real time mass spectrometry (DART-MS). Taken together, these data indicate that addition of the tetrazine to 1 results in the expected click reaction and initiates self-immolation of the thiocarbamate moiety, thus producing COS.
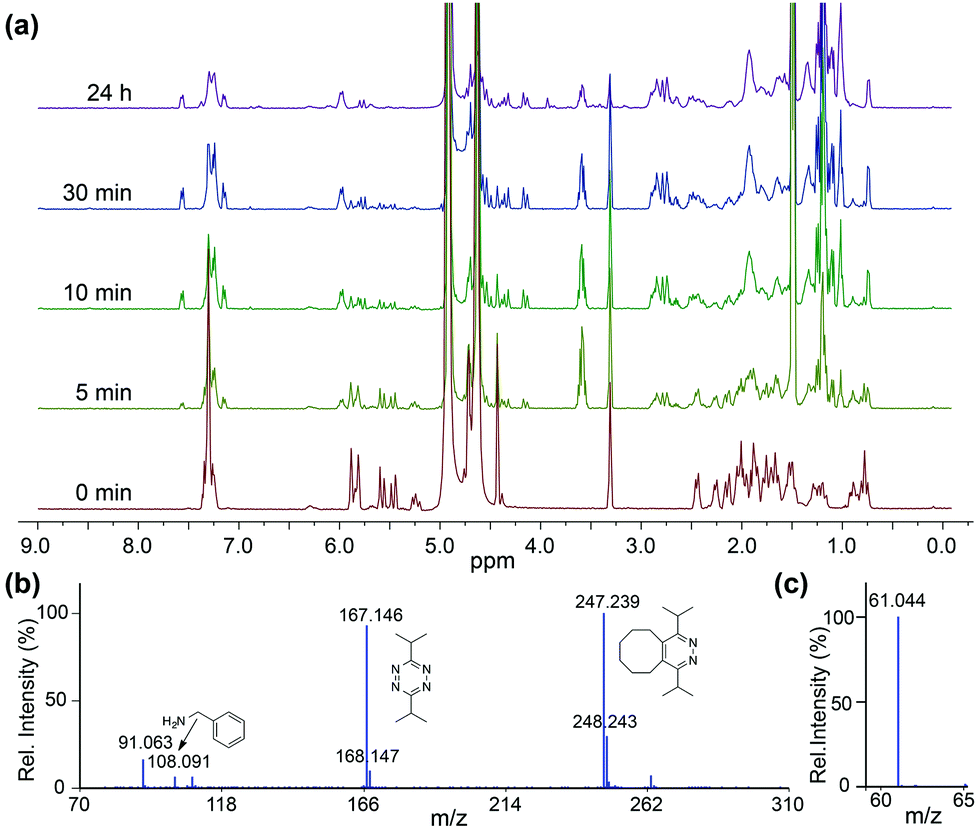 | ||
| Fig. 1 (a) 1H NMR spectra of the reaction of 1 and tetrazine (b) ESI-MS of reaction products, confirming self-immolation. (c) MS confirmation of COS formation. | ||
Having confirmed the fidelity of the IEDDA reaction, we next investigated click-and-release H2S-donation from this system in aqueous buffer at physiological pH (PBS, pH 7.4) using an H2S-selective electrode (Fig. 2). Non-enzymatic background hydrolysis of COS to H2S is very slow at physiological pH, but is rapid in the presence of carbonic anhydrase (CA). Using biologically-relevant CA concentrations (25 μg mL−1) we first monitored TCO 1 alone and confirmed that H2S is not released spontaneously in the presence of CA. As anticipated, the bis-isopropyl-1,2,4,5-tetrazine alone also failed to produce an H2S response. We next monitored H2S release from TCO 1 (50 μM) with varying concentrations of tetrazine (5–25 equiv.) and observed increased H2S production in the presence of excess tetrazine. Using a calibration curve, we measured 12 μM H2S release from 50 μM TCO 1 with 25 equiv. of tetrazine, resulting in an H2S release efficiency of approximately 25%. As additional confirmation of the importance of CA for H2S formation, we performed identical reactions in the presence of acetazolamide (AAA, 2.5 μM), a known CA inhibitor. Furthermore, use of the control compound TCO 2 in the presence of excess tetrazine failed to generate COS/H2S. Together, these data confirm that the IEDDA click reaction is necessary to generate COS, and that uninhibited CA is required for efficient conversion of this released COS to H2S at physiological pH.
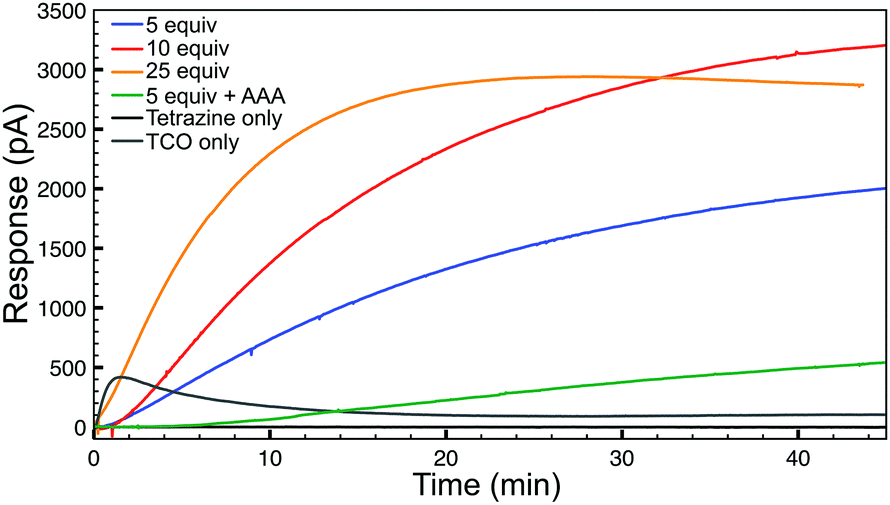 | ||
| Fig. 2 H2S release profiles from TCO 1 (50 μM) with 5–25 equiv. of tetrazine in the presence of CA (25 μg mL−1) in buffer (PBS, pH 7.4). | ||
To demonstrate the basic biological compatibility of the reaction, we also investigated H2S release from TCO 1 (50 μM) with the tetrazine (500 μM) in complex media (Fig. 3). For these experiments, we chose to use whole sheep and bovine blood due to the presence of CA. Using sheep blood and plasma, diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in PBS (pH 7.4) with no additional CA added, a similar H2S release profile was observed using an H2S-selective electrode. Additionally, H2S production was also observed in diluted whole bovine blood, although the process was slower. These experiments confirm that bio-orthogonal click-and-release strategy has significant potential within a biological environment and endogenous CA levels are sufficient to allow for H2S donation from the released COS. Additionally, we confirmed the cellular compatibility of TCO 1 using the CCK-8 cell viability assay, which indicated that concentrations up to 100 μM of TCO 1 are not cytotoxic in N2A neuroblastoma cells (see ESI†).
:1 in PBS (pH 7.4) with no additional CA added, a similar H2S release profile was observed using an H2S-selective electrode. Additionally, H2S production was also observed in diluted whole bovine blood, although the process was slower. These experiments confirm that bio-orthogonal click-and-release strategy has significant potential within a biological environment and endogenous CA levels are sufficient to allow for H2S donation from the released COS. Additionally, we confirmed the cellular compatibility of TCO 1 using the CCK-8 cell viability assay, which indicated that concentrations up to 100 μM of TCO 1 are not cytotoxic in N2A neuroblastoma cells (see ESI†).
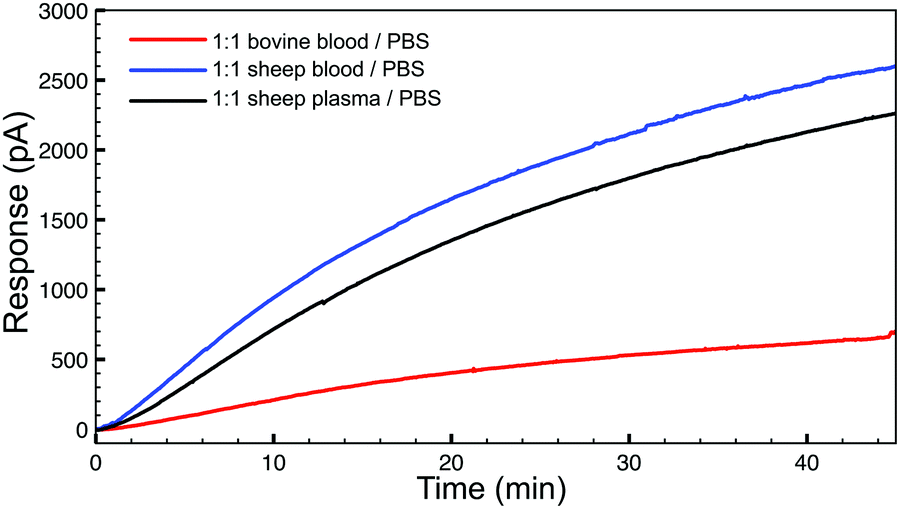 | ||
| Fig. 3 H2S release profiles from TCO 1 (50 μM) with 10 equiv. of tetrazine in whole bovine blood (red), whole sheep blood (blue), and sheep plasma (grey), diluted 1:1 with buffer (PBS, pH 7.4). | ||
In an effort to expand this strategy to a cellular environment, we attempted to obtain cell images using a variety of fluorescent probes for H2S, including HSN2, WSP-5, and SF7-Am.17–19 Unfortunately, we found that the click-and-release reaction was not compatible with these current fluorescent detection strategies for H2S. This observation was confirmed in cuvette-based fluorimetry studies as well, in which no fluorescent turn-on was observed after several hours despite the production of H2S, as confirmed by H2S-electrode experiments. Although unexpected, this outcome may be due to slower and/or less-efficient COS/H2S release from this first-generation IEDDA platform than from previously reported COS/H2S donors. In a closed system, it is also possible that the tetrazine may also scavenge the generated H2S, as evidenced by a recent report demonstrating that H2S can partially reduce dialkoxy tetrazines to the dihydrotetrazine.20 Therefore, future investigations into the differential reactivity of H2S with substituted tetrazines appears warranted, both to increase the biocompatibility in this system and also to increase the initial efficiency of the IEDDA click reaction.21 For example, a recent report highlighted that the efficiency of the IEDDA reaction can be improved through strategic choice of the tetrazine. These, as well as other modifications to the thiocarbamate scaffold are expected to provide much more efficient H2S release from future click-and-release scaffolds.
In summary, we have reported the first example of COS/H2S donors activated by a bio-orthogonal trigger, which provides a significant step toward developing controllable H2S donors with high temporal resolution. Given the novelty of this bio-orthogonal reaction in the field of sulfide donation, as well as the significant impact that similar click strategies have provided to adjacent fields in chemical biology, we anticipate that future optimization of this system will result in fast and highly targeted methods for H2S donation.
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1309047. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. Support was also provided by the Sloan Foundation and Dreyfus Foundation (to MDP); and the Research Foundation of the New York State (to MR).
References
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- Z. J. Song, M. Y. Ng, Z.-W. Lee, W. Dai, T. Hagen, P. K. Moore, D. Huang, L.-W. Deng and C.-H. Tan, MedChemComm, 2014, 5, 557–570 RSC.
- K. R. Olson, Antioxid. Redox Signaling, 2012, 17, 32–44 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef CAS PubMed.
- C. R. Powell, J. C. Foster, B. Okyere, M. H. Theus and J. B. Matson, J. Am. Chem. Soc., 2016, 138, 13477–13480 CrossRef CAS PubMed.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638–14642 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4762–4767 CrossRef CAS PubMed.
- R. Rossin, P. R. Verkerk, S. M. van den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed., 2010, 49, 3375–3378 CrossRef CAS PubMed.
- J. M. Mejia Oneto, I. Khan, L. Seebald and M. Royzen, ACS Central Sci., 2016, 2, 476–482 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- R. Rossin, S. M. J. van Duijnhoven, W. ten Hoeve, H. M. Janssen, L. H. J. Kleijn, F. J. M. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697–1706 CrossRef CAS PubMed.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS PubMed.
- X. Ji, C. Zhou, K. Ji, R. E. Aghoghovbia, Z. Pan, V. Chittavong, B. Ke and B. Wang, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201608732.
- L. A. Montoya and M. D. Pluth, Chem. Commun., 2012, 48, 4767–4769 RSC.
- B. Peng, W. Chen, C. R. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem. – Eur. J., 2014, 20, 1010–1016 CrossRef CAS PubMed.
- V. S. Lin, A. R. Lippert and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7131–7135 CrossRef CAS PubMed.
- Z. Zhao, L. Cao, T. Zhang, R. Hu, S. Wang, S. Li, Y. Li and G. Yang, ChemistrySelect, 2016, 1, 2581–2585 CrossRef CAS.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. C. Ngai, Z. Lin, S. Zheng, J. Wang, J. Zhao, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2016, 55, 14046–14050 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and experimental details. See DOI: 10.1039/c6cc09547j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |