
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen bonds between methanol and the light liquid olefins 1-pentene and 1-hexene: from application to fundamental science†
Zhaoxi
Zhang
a,
Tiancun
Xiao
*a,
Hamid
Al-Megren
b,
Saud A.
Aldrees
b,
Mohammad
Al-Kinany
b,
Vladimir L.
Kuznetsov
a,
Maxim L.
Kuznetsov
 c and
Peter P.
Edwards
*a
c and
Peter P.
Edwards
*a
aKing Abdulaziz City for Science and Technology – Oxford Centre of Excellence in Petrochemicals (KOPRC), Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, OX1 3QR, UK. E-mail: peter.edwards@chem.ox.ac.uk; xiao.tiancun@chem.ox.ac.uk
bMaterials Research Institute, King Abdulaziz City for Science and Technology, P.O. Box 6086, Riyadh, 11442, Saudi Arabia
cCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: max@mail.ist.utl.pt
First published on 9th March 2017
Abstract
We have recently developed a new extraction process for significantly reducing the olefin content in commercial FCC gasoline. To gain insights into the origins of this process, we have investigated the dissolution of the light liquid olefins 1-pentene and 1-hexene in methanol through computer modelling together with NMR spectroscopy. We find two important hydrogen bonding modes for methanol olefin interactions – namely, O–H⋯π and C–H⋯O.
Fluid catalytic cracking (FCC) is one of the principal oil refining processes which converts heavy fractions crude oil into transportation fuels. Unfortunately, as-produced FCC gasoline contains high levels of olefins (in some cases up to 59% v/v) and appreciable levels of thiophenic sulphur impurities and aromatics,1–4 all of which require further treatment to attain acceptable environmental levels set by the World Fuel Charter Standard.5 The olefin and sulphur content is conventionally reduced by hydrogenation and hydrodesulphurization but inevitable olefin saturation leads to fuel octane loss and excessive consumption of hydrogen (produced of course by the carbon-footprint-intensive steam-reforming of methane).2,6
We have recently developed7 a new “Extractive Refining” (ER) process which uses methanol as a highly effective solvent to extract olefins and other products such as organic sulphur compounds (OSCs) from FCC gasoline (Fig. 1: see ESI† for experiment details). For example, after just one methanol extraction cycle, the level of total olefin in FCC gasoline, which consists of C4–C8 olefins,8,9 were significantly reduced by some 31.6% (Fig. 1). The resultant mixture of the olefin containing methanol can also be catalytically converted into high quality gasoline and other products.
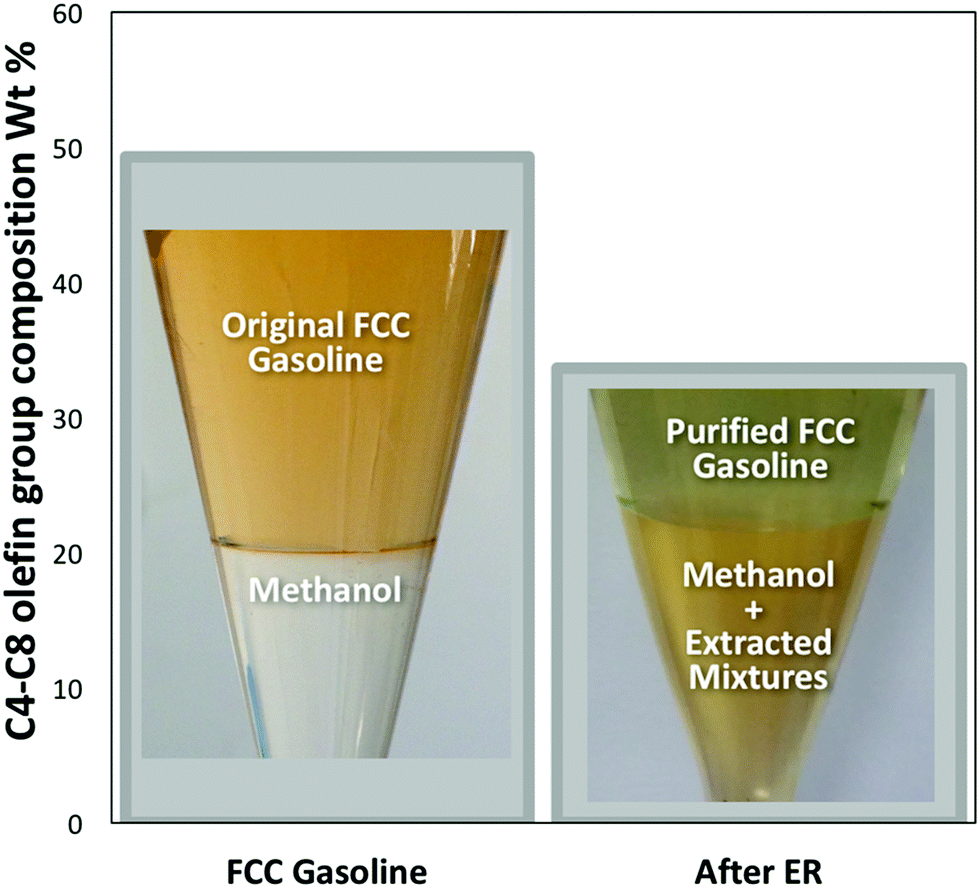 | ||
| Fig. 1 Commercial FCC gasoline (SINOPEC) composition of C4–C8 olefins both before and after one cycle of the Extractive Refining process. | ||
We targeted methanol as a selective solvent for extraction since it is a high-volume, inexpensive commodity chemical, readily produced from a range of carbonaceous feedstocks, including biomass and of course CO2, itself.10,11
To enhance the efficiency of the new ER process and with that the maximization of the environmental gain, it is necessary to have a fundamental understanding of the interaction, for example, between the extracting solvent, methanol and the constituent solute olefins.
It has been shown that C4–C8 olefins in exhaust emissions from vehicles are mainly unburnt components,8,9 while C6 olefins are the major olefinic constituent in FCC gasoline fraction.12,13 We therefore targeted both the C5 and C6 homologous members for this study. Importantly, we find that both these light liquid olefins, 1-pentene and 1-hexene are completely soluble in methanol at room temperature and room pressure.
It was previously reported that 1-heptene is mutually soluble in methanol above the Upper Critical Solution Temperature of 285 K and 271.6 K, respectively.14–16 However, these studies did not interrogate the nature of the light liquid olefin–methanol interaction. Recently Heger et al. investigated the hydrogen bond between methanol and ethene molecules in the gas phase,17 Oku et al. proposed a structure for the methanol–2-butene complex and structures based on their quantum chemical calculation of optimised interaction energies, but no direct experimental evidence was presented.18 Medel et al. also investigated the molecular docking via olefinic O–H⋯π interactions,19 but these studies did not encompass possible hydrogen bond interactions between methanol and liquid olefins, and the effect of these interactions on the solubility of olefins in methanol. Zwier extensively reviewed the complexation of methanol and the π-system of molecular benzene in detail, but did not extend the study to interactions between liquid methanol and olefin, the present system of interest here.20
To explore the possible origins of the interaction between the prototypical light olefins and liquid methanol we first carried out quantum chemical calculations of 1-pentene and 1-hexene (alpha-olefins are the main olefins in FCC gasoline) with a single methanol molecule (olefin⋯methanol) and found the stable structure of these olefins surrounded by 8 methanol molecules (olefin⋯8 methanol, see ESI† for full computational details). In the most stable equilibrium structures of the olefin⋯methanol associates, the hydroxyl hydrogen atom of methanol interacts with the π (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double) bond of the olefin with the OH⋯(CC)midpoint distances of 2.36–2.41 Å (Table 1, Fig. 2 and Table S2, Fig. S1 in ESI†).
C double) bond of the olefin with the OH⋯(CC)midpoint distances of 2.36–2.41 Å (Table 1, Fig. 2 and Table S2, Fig. S1 in ESI†).
| 1-Hexene⋯methanol | 1-Hexene⋯8 methanola,c | |||
|---|---|---|---|---|
| Gas phasea | SMD (1-hexene as solvent)b | SMD (methanol as solvent)b | ||
|
a Isolated molecular associate without bulk solvent effects.
b SMD solvation model.
c Molecular associate with a 1-hexene molecule surrounded by 8 methanol molecules.
d Binding energies (in kJ mol−1) at the M06-2X/6-311+G** level.
e BSSE corrected value.
f Binding energies (in kJ mol−1) at the CCSD(T)/6-311+G**//M06-2X/6-311+G** level.
g Intermolecular OH⋯(CC)midpoint distances (in Å).
h Electron density (in e Å−3) at the OH⋯(CC) BCP.
i Laplacian (in e Å−5) at the OH⋯(CC) BCP.
j Energy density at the OH⋯(CC) BCP (in Hartree Å−3).
|
||||
| E b | −19.6 (−17.2)e | −19.8 | −13.6 | |
| E b | −15.8 | −14.8 | −11.3 | |
| l(OH⋯π)g | 2.409 | 2.376 | 2.360 | 2.361 |
| ρ | 0.082 | 0.090 | 0.088 | 0.096 |
| ∇2ρi | 0.859 | 0.912 | 0.919 | 1.012 |
| H b | 0.010 | 0.011 | 0.012 | 0.010 |
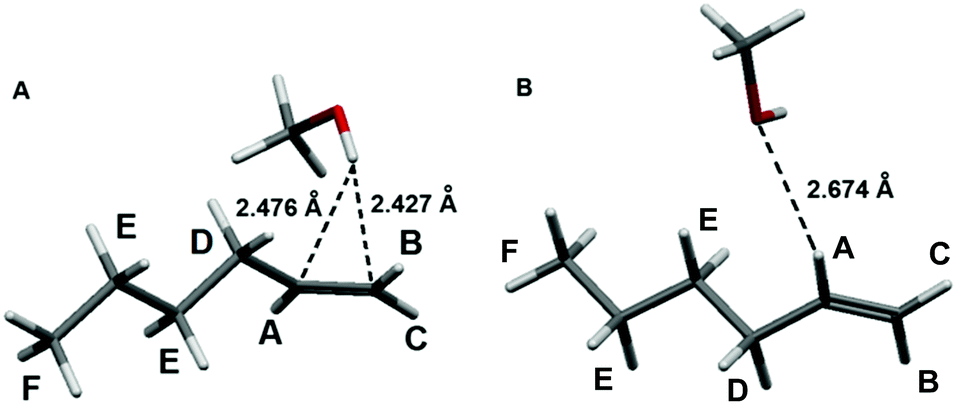 | ||
| Fig. 2 Equilibrium structures of the 1-hexene⋯methanol associates exhibiting the O–H⋯(CC) (A), the most stable structure and C–H⋯O (B) interactions. | ||
The methanol⋯1-hexene (O–H⋯π) binding energies computed at the M06-2X and CCSD(T) levels, both with and without basis set superposition error (BSSE) correction, are given in Table 1. Their values (−11.3 to −19.8 kJ mol−1) are comparable for both levels, and the BSSE correction is rather insignificant (−2.4 kJ mol−1). The topological analysis of the electron density distribution (the AIM method) revealed the existence of a bond critical point (BCP) corresponding to the interaction between the methanol hydroxyl hydrogen atom and the π bond in the 1-hexene⋯methanol associate. The electron density values (ρ) at this BCP do not exceed 0.10 e Å−3 (Table 1), the electron density Laplacian (∇2ρ) is positive and the energy density values (Hb) are slightly positive. All these values are typical for the weak hydrogen bonding.21–23 The natural bond orbital analysis demonstrates very small charge transfer (0.003 e) from 1-hexene to the methanol molecule in the associate 1-hexene⋯methanol which is associated with the π (CC) → σ*(OH) transition with the E(2) second order NBO energy of 10.5 kJ mol−1. Both models, i.e. 1-hexene⋯methanol cluster with bulk solvent effects and 1-hexene⋯8 methanol cluster with explicit first solvation shell around olefin molecule, demonstrate very similar bonding features. Results obtained for the 1-pentene + methanol system are similar and given in ESI.†
These calculations indicate that the dissolution process could involve hydrogen bonds formed between the olefin and methanol, via O–H⋯π and C–H⋯O interactions as we will illustrate experimentally.18,24
To further probe the nature of the interaction of hydroxyl group in methanol with this light olefin, the mixtures were studied by high resolution 1H-NMR spectroscopy using a Bruker Advance III HD Nanobay 400 MHz NMR spectrometer and utilising deuterated methanol and cyclohexane (to render these solvents “invisible” in the H spectra). Cyclohexane was chosen as the reference solvent in this analysis, and it is also mutually soluble with the olefin. Tetramethylsilane (TMS) was used as the internal standard, which is assigned the chemical shift zero (0 ppm).
We determined the precision and standard error of the chemical shifts by careful fitting of the NMR line positions from typically 10 separate runs for each sample and is ±0.005 ppm. The 1H-NMR spectra of solutions of 1-pentene and 1-hexene in both of these solvents are shown in Fig. 3 and 4.
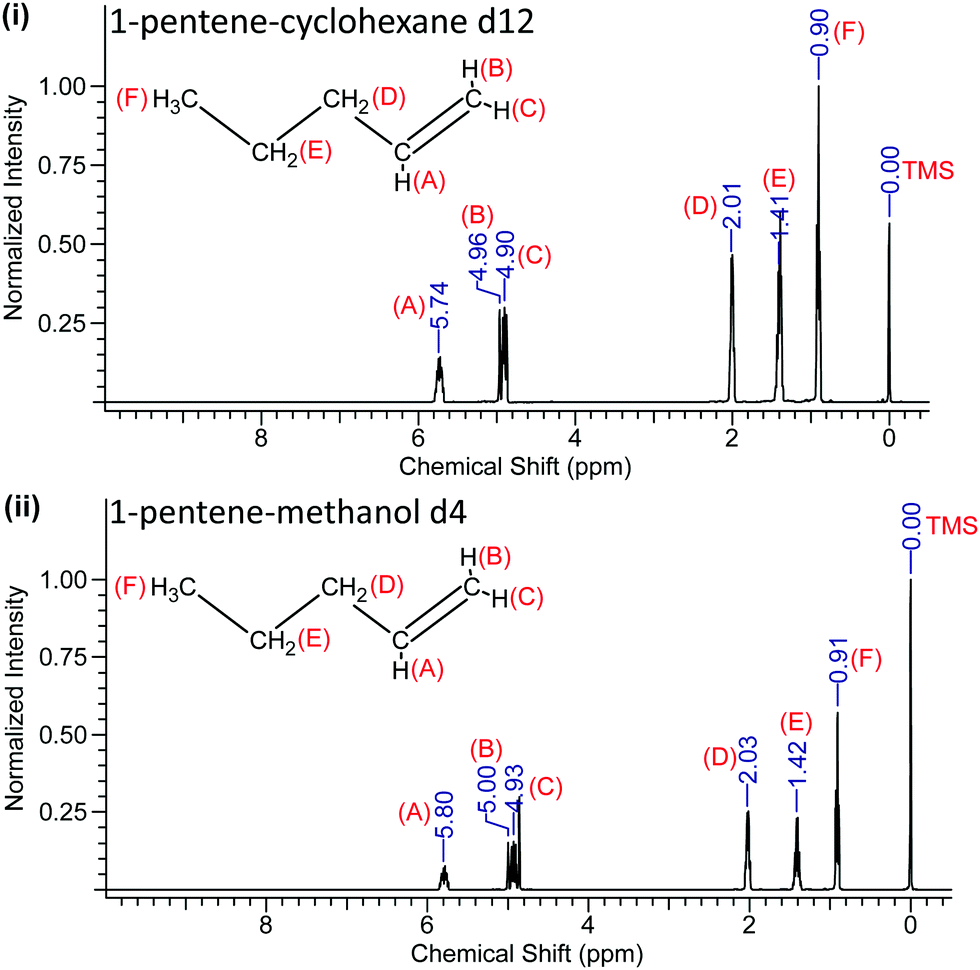 | ||
| Fig. 3 1H-NMR spectra of 1-pentene in cyclohexane-d12 (i) and in methanol-d4 (ii). | ||
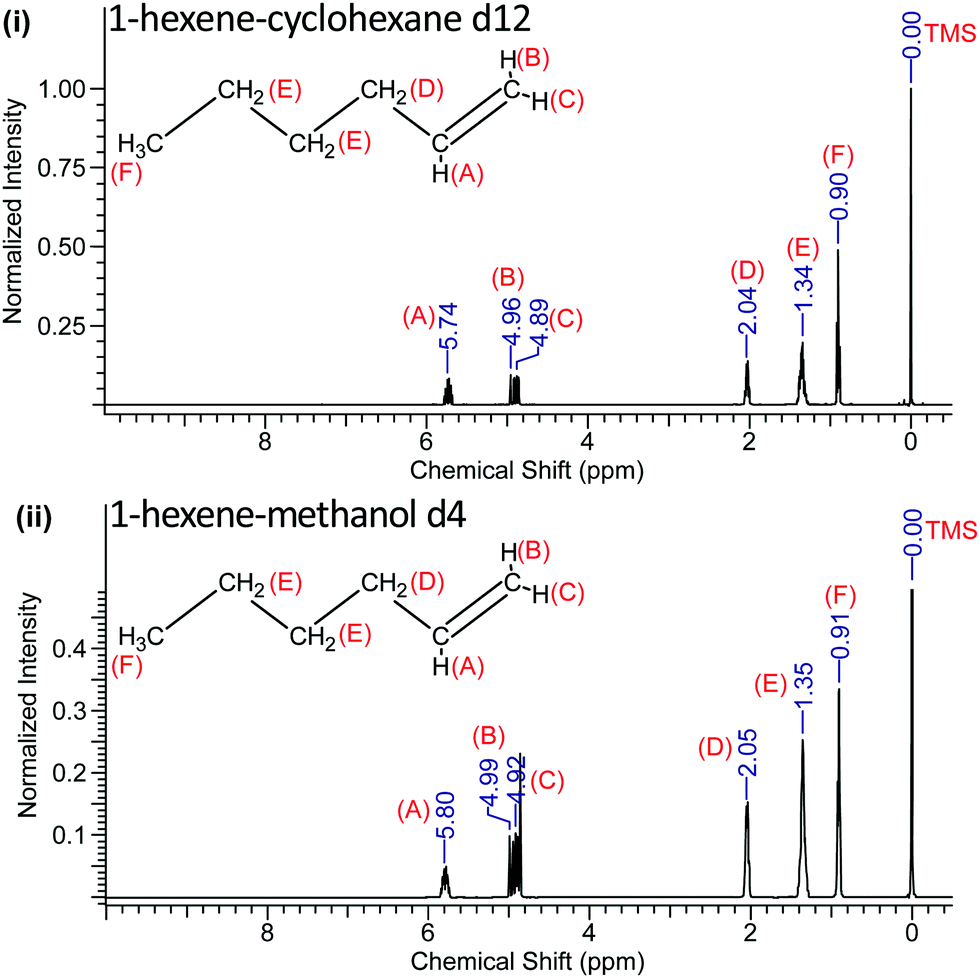 | ||
| Fig. 4 1H-NMR spectra of 1-hexene in cyclohexane-d12 (i) and in methanol-d4 (ii). | ||
Here 1-pentene and 1-hexene are regarded as the solutes and methanol as the host solvent. The resulting chemical shifts are a direct consequence of electron redistribution around H atoms following H bond formation. In pioneering work, Buckingham et al. defined four interactions responsible for solvent effects in the NMR of solute species,25 hydrogen bonding, anisotropy of the solute and solvent interactions and dipole–dipole and van der Waals interactions. The quantitative treatment of these relative contributions to solvent shifts of solutes still remains a major challenge.25,26
The recorded values of the NMR chemical shifts of the various protons are indicated in Fig. 3 and 4 and the location (A–F) of the 1-pentene and 1-hexene's protons are also labelled.
The NMR data reveal that when 1-pentene and 1-hexene are dissolved in methanol, all the protons are de-shielded relative to those in cyclohexane solutions with chemical shifts moving downfield. The protons located at A, B and C become more de-shielded as compared to the other protons located at D, E and F (Fig. 3 and 4). The chemical shift changes for the 1-pentene's proton located at (A), (B) and (C) were 0.06 ppm, 0.04 ppm and 0.03 ppm, for the 1-hexene's proton located at (A), (B) and (C) were 0.06 ppm, 0.03 ppm and 0.03 ppm (Table 2). It is important to note that these shifts are outside of the experimental error estimates (±0.005 ppm) in carefully measured chemical shifts across ten recordings of each sample and the results reveal excellent reproducibility.27 We note that these (A), (B) and (C) protons are located on the unsaturated carbon atom (Fig. 3 and 4).
| 1H chemical shifts (experimental) δa | 1H chemical shifts (calculated) δ | |||||
|---|---|---|---|---|---|---|
| Proton | 1-pen in C-d12 | 1-pen in M-d4 | 1-hex in C-d12 | 1-hex in M-d4 | 1-hex | 1-hex⋯MeOH |
| a Chemical shift (in ppm), standard errors are ±0.005 ppm. b Average values for all protons of a given type. | ||||||
| A | 5.74 | 5.80 | 5.74 | 5.80 | 6.72 | 7.05 |
| B | 4.96 | 5.00 | 4.96 | 4.99 | 5.80 | 5.91 |
| C | 4.90 | 4.93 | 4.89 | 4.92 | 5.60 | 5.74 |
| D | 2.01 | 2.03 | 2.04 | 2.05 | 2.09b | 2.26b |
| E | 1.41 | 1.42 | 1.34 | 1.35 | 1.26b | 1.30b |
| F | 0.90 | 0.91 | 0.90 | 0.91 | 0.96b | 0.97b |
The complete 1H-NMR results for the mixtures of 1-pentene and 1-hexene in cyclohexane-d12 or methanol-d4 are listed in Table 2.
Bulk magnetic susceptibility differences between cyclohexane and methanol will make contributions to the magnetic shielding of all constituent protons; and these should be identical and independent of their position along the hydrocarbon chain. The fact that the observed chemical shifts for the H associated with all constituent carbon atoms are different in magnitude (Table 2) reflect site-specific interactions which contribute to the magnetic shielding of individual protons.
The combination of the spectroscopic and computational data enables us to conclude that these NMR chemical shifts originate from the two types of olefin–methanol interactions highlighted in our computational studies (Fig. 2).
The first is the O–H⋯π interaction,17,18 sometimes referred to as the “pi hydrogen bond”. When the O–H⋯π hydrogen bond is formed, the 1-pentene or 1-hexene acts as a π base (hydrogen bond acceptor) where the electron density from the olefin's π-electron system is donated to the proton (σ* orbital) of the methanol's hydroxyl, and this reduces the electron density of the protons (A), (B) and (C) which are linked with the unsaturated carbon atom in the olefins.22,23,28,29
The second type is the C–H⋯O interaction,24 where the oxygen atom on methanol acts as an hydrogen bond acceptor,22,23 and due to an electrostatic interaction the electron density from the methanol's oxygen atom is donated to the protons located at (B) and (C) of 1-pentene or 1-hexene.30–32 However, compared with the energy of O–H⋯π interactions, the C–H⋯O interaction between these olefins and the methanol molecule is weaker.18
Because of the formation of these hydrogen bonds, the electron density of the protons linked to the unsaturated carbon atom is thereby decreased. This leads to the reduction of the shielding effect on these protons. It is particularly noteworthy that the chemical shift of the protons (A) to (C) increases more than the remaining protons (D) to (F) (Table 2). In addition, proton chemical shift of 1-hexene and 1-hexene in methanol calculated from DFT shows the similar trend (Table 2).
Hence, through the analysis of these 1H-NMR results and computational studies, we conclude that hydrogen bonds are formed (1) between methanol and olefins through the interaction of the π-electron cloud of 1-pentene or 1-hexene and the hydrogen atom of the methanol hydroxyl group,18,33 together with the interaction of a proton of these olefins and the oxygen atom of the hydroxyl group on methanol.31
In conclusion, we have demonstrated that both 1-pentene and 1-hexene are completely miscible in methanol at room temperature. A combination of computational and high resolution NMR studies indicates that the interaction between these olefins and the host solvent methanol molecules is mainly due to the presence of O–H⋯π and C–H⋯O hydrogen bonds. Understanding the microscopic interactions of these prototypical light liquid olefins with methanol provides valuable insights into this new removal/separation process for olefin reduction from FCC gasoline in refineries world-wide.
We thank KACST Saudi Arabia and EPSRC for financial support, Dr Jamie Ferguson for constructive discussions and Dr Karl Harrison for assembling Fig. 1. Thanks are due to Dr Nick Rees for NMR measurement and expertise and Professor Robert K. Thomas for his interest. We thank SINOPEC for providing us with FCC gasoline samples. This work has been partially supported by the Foundation for Science and Technology (FCT, Portugal) (project UID/QUI/00100/2013).
Notes and references
- R. Harding, A. Peters and J. Nee, Appl. Catal., A, 2001, 221, 389 CrossRef CAS.
- J. Liu, S. Zhao, X. Chen and B. Shen, Fuel, 2016, 166, 467 CrossRef CAS.
- W. Yinhui, Z. Rong, Q. Yanhong, P. Jianfei, L. Mengren, L. Jianrong, W. Yusheng, H. Min and S. Shijin, Fuel, 2015, 166, 543 CrossRef.
- F. Pradelle, S. L. Braga, A. R. F. A. Martins, F. Turkovics and R. N. C. Pradelle, Energy Fuels, 2015, 29, 7753 CrossRef CAS.
- R. C. Pereira and V. Pasa, Fuel, 2006, 85, 1860 CrossRef CAS.
- A. Daudin, A. F. Lamic, G. Perot, S. Brunet, P. Raybaud and C. Bouchy, Catal. Today, 2008, 130, 221 CrossRef CAS.
- H. Al-Megren, M. Al-Kinany, S. Aldrees, V. L. Kuznetsov, P. P. Edwards, Z. Zhang and T. Xiao, Extraction of alkenes, WO. Pat., WO2015177531 A1, 2015 Search PubMed.
- J. Bailey and S. Eggleston, Sci. Total Environ., 1993, 134, 263 CrossRef CAS.
- O. Ramnäs, U. Östermark and G. Petersson, Chromatographia, 1994, 38, 222 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636 CrossRef CAS PubMed.
- W. Kiatkittipong, S. Wongsakulphasatch, N. Tintan, N. Laosiripojana, P. Praserthdam and S. Assabumrungrat, Fuel Process. Technol., 2011, 92, 1999 CrossRef CAS.
- Z. Rusong, G. Junbin and Z. Juanjuan, Petrochem. Technol., 2009, 3, 9 Search PubMed.
- A. W. Francis, Ind. Eng. Chem., 1944, 36, 764 CrossRef CAS.
- O. P. Pavlova, A. A. Gaile, V. A. Proskuryokov and I. F. Li, Zh. Fiz. Khim., 1975, 49, 2874 CAS.
- Alcohols With Hydrocarbons, IUPAC Solubility Data Series, ed. D. Shaw, A. Skrzecz, J. W. Lorimer and A. Maczunski, Oxford University Press, Oxford, 1994, vol. 56 Search PubMed.
- M. Heger, R. A. Mata and M. A. Suhm, Chem. Sci., 2015, 6, 3738 RSC.
- K. Oku, H. Watanabe, M. Kubota, S. Fukuda, M. Kurimoto, Y. Tsujisaka, M. Komori, Y. Inoue and M. Sakurai, J. Am. Chem. Soc., 2003, 125, 12739 CrossRef CAS PubMed.
- R. Medel, M. Heger and M. A. Suhm, J. Phys. Chem. A, 2014, 119, 1723 CrossRef PubMed.
- T. S. Zwier, Annu. Rev. Phys. Chem., 1996, 47, 205 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154 CrossRef CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, Pure Appl. Chem., 2011, 83, 1637 CAS.
- D. J. Sutor, Nature, 1962, 195, 68 CrossRef CAS.
- A. D. Buckingham, T. Schaefer and W. G. Schneider, J. Chem. Phys., 1961, 34, 1064 CrossRef CAS.
- R. J. Abraham, J. J. Byrne, L. Griffiths and M. Perez, Magn. Reson. Chem., 2006, 44, 491 CrossRef CAS PubMed.
- R. E. Hoffman, Magn. Reson. Chem., 2006, 44, 606 CrossRef CAS PubMed.
- D. E. Rosenfeld, Z. Gengeliczki and M. D. Fayer, J. Phys. Chem. B, 2009, 113, 13300 CrossRef CAS PubMed.
- I. V. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons Ltd, Chichester, 2016 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 2001 Search PubMed.
- B. M. Kariuki, K. D. M. Harris, D. Philp and J. M. Robinson, J. Am. Chem. Soc., 1997, 119, 12679 CrossRef CAS.
- G. R. Desiraju, Chem. Commun., 2005, 2995 RSC.
- K. Kowski, W. Lüttke and P. Rademacher, J. Mol. Struct., 2001, 567, 231 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, table with atomic coordinates of the calculated structures. See DOI: 10.1039/c6cc09545c |
| This journal is © The Royal Society of Chemistry 2017 |