
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homoleptic organolanthanide compounds supported by the bis(dimethylsilyl)benzyl ligand†
Kasuni C.
Boteju
 ,
Arkady
Ellern
and
Aaron D.
Sadow
*
,
Arkady
Ellern
and
Aaron D.
Sadow
*
Department of Chemistry and US DOE Ames Laboratory, Iowa State University, Ames IA 50011, USA. E-mail: sadow@iastate.edu
First published on 19th December 2016
Abstract
A β-SiH functionalized benzyl anion [C(SiHMe2)2Ph]− is obtained by deprotonation of HC(SiHMe2)2Ph with KCH2Ph or by reaction of KOtBu and (Me2HSi)3CPh; LnI3(THF)n and three equivalents of this carbanion combine to provide homoleptic tris(alkyl)lanthanide compounds Ln{C(SiHMe2)2Ph}3 (Ln = La, Ce, Pr, Nd) containing secondary metal–ligand interactions.
Synthesis of homoleptic organolanthanide complexes, particularly those of the early trivalent lanthanides (La–Nd), is challenging due to the large radii of these elements, polar bonding, high charge, and high Lewis acidity.1 Such homoleptic compounds should be valuable for the synthesis of new catalysts and new materials,2 yet solvent- or donor-group-free, salt-free, and thermally robust organolanthanide compounds are not readily accessed for the larger metal centers. For example, the reaction of MeLi and LaCl3 gives Li3[LaMe6] as a TMEDA adduct.3 Three THF molecules coordinate to the labile tris(benzyl)lanthanum allowing isolation of LaBn3(THF)3,4,5 however, even this adduct eliminates toluene at room temperature with a half-life of ca. 2 h. The persistence of related compounds may be enhanced by chelating benzylic ligands, for example Ln(CH(NMe2)Ph)36 or Ln(CH2C6H4-2-NMe2)3.7 An alternative approach combines bulky β-SiMe3 with the benzyl group in C(SiMe3)2Ph,8 exemplified by a bis(alkyl)calcium compound possessing metal–aryl π-interactions. Coordinative unsaturation is important to the reactivity of organolanthanides, and donors such as TMEDA or THF can diminish reactivity,9 facilitate alkane elimination,10 or react by C–O bond cleavage.11 While donor-free lanthanum and cerium compounds such as Ln{CH(SiMe3)2}3 are known, they are inconveniently accessed through multistep synthesis via Ln{O(2,6-C6H3tBu2)}3.12,13
A strategy for stabilizing coordinatively unsaturated rare earth amides has involved the incorporation of SiH groups, which form labile secondary interactions with the lanthanide center.14 Furthermore, the SiH moiety provides a powerful signature in 1H and 29Si NMR and IR spectra. This β-SiH strategy may also be applied to alkyls, and the ligand C(SiHMe2)3 supports trivalent yttrium and divalent ytterbium and samarium homoleptic alkyls containing secondary Ln↼H–Si interactions.15,16 Recently, we reported Ce{C(SiHMe2)3}3 as a precursor to a zwitterionic hydrosilylation catalyst.17 New chemistry might be accessed with alkyl ligand variations that include both β-SiH and benzylic functionalities, and these groups could compete to enhance the homoleptic compounds' resistance to undesired ligand elimination pathways. Both SiH and benzyl groups may have significant charge delocalization and secondary interactions that might stabilize homoleptic compounds. A single ligand containing both elements, namely –C(SiHMe2)2Ph, would test these ideas. Here we report the synthesis of alkane precursors, two routes to potassium alkyl reagents, and isolation and characterization of a series of homoleptic organolanthanide complexes.
Reductive coupling of HCPhBr2 and ClSiHMe2 affords HC(SiHMe2)2Ph (1; eqn (1)) on preparative scale.
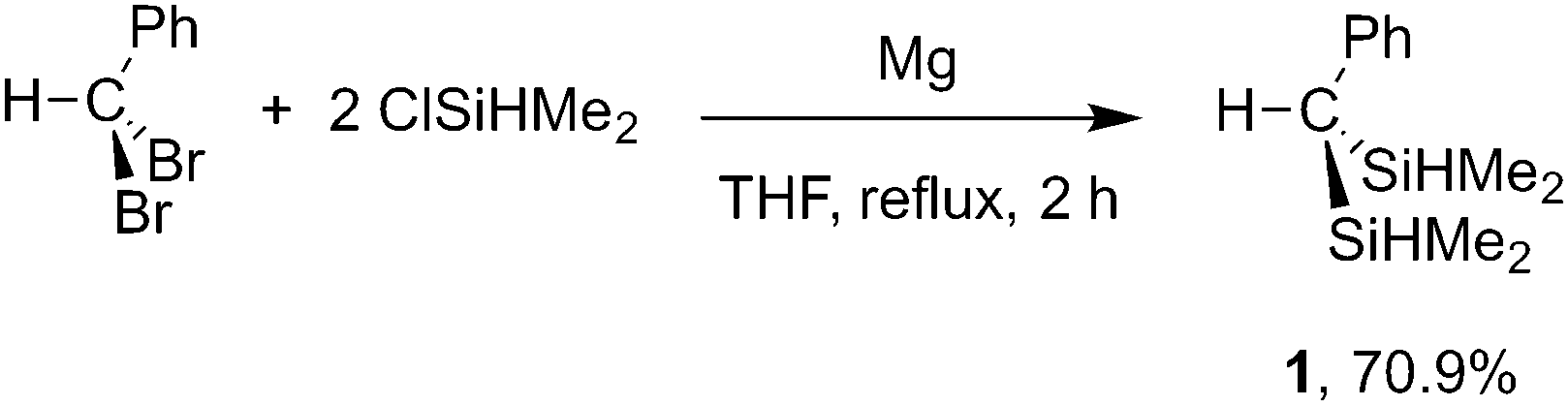 | (1) |
While HC(SiHMe2)318 reacts readily with lithium diisopropylamide,19 deprotonation of HC(SiHMe2)2Ph is more challenging. Attempts to synthesize [C(SiHMe2)2Ph]− using LiN(SiMe3)2, nBuLi, KH, or KC(SiHMe2)3 as bases returned HC(SiHMe2)2Ph. Potassium benzyl (KBn) gives Me2SiBn2 as the major product in its reaction with 1 at room temperature. Fortunately, reactions with KBn performed at −78 °C yielded a mixture now dominated by KC(SiHMe2)2Ph (2), assigned to a doublet at 0.47 ppm in the 1H NMR spectrum. This signal was affected by addition of TMEDA, which gave a new doublet at 0.57 ppm. In preparative scale reactions, the desired potassium alkyl is crystallized from pentane at −30 °C to provide Ph(Me2HSi)2CK(TMEDA) (2·TMEDA) as dark red crystals (eqn (2)), albeit in low isolated yield.
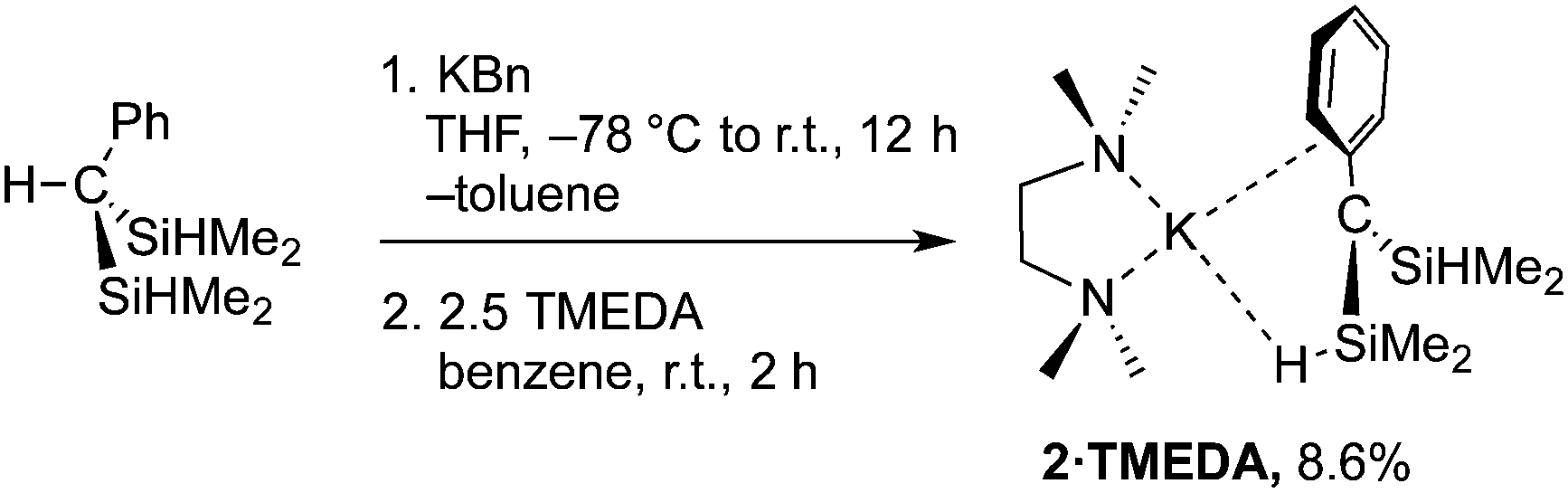 | (2) |
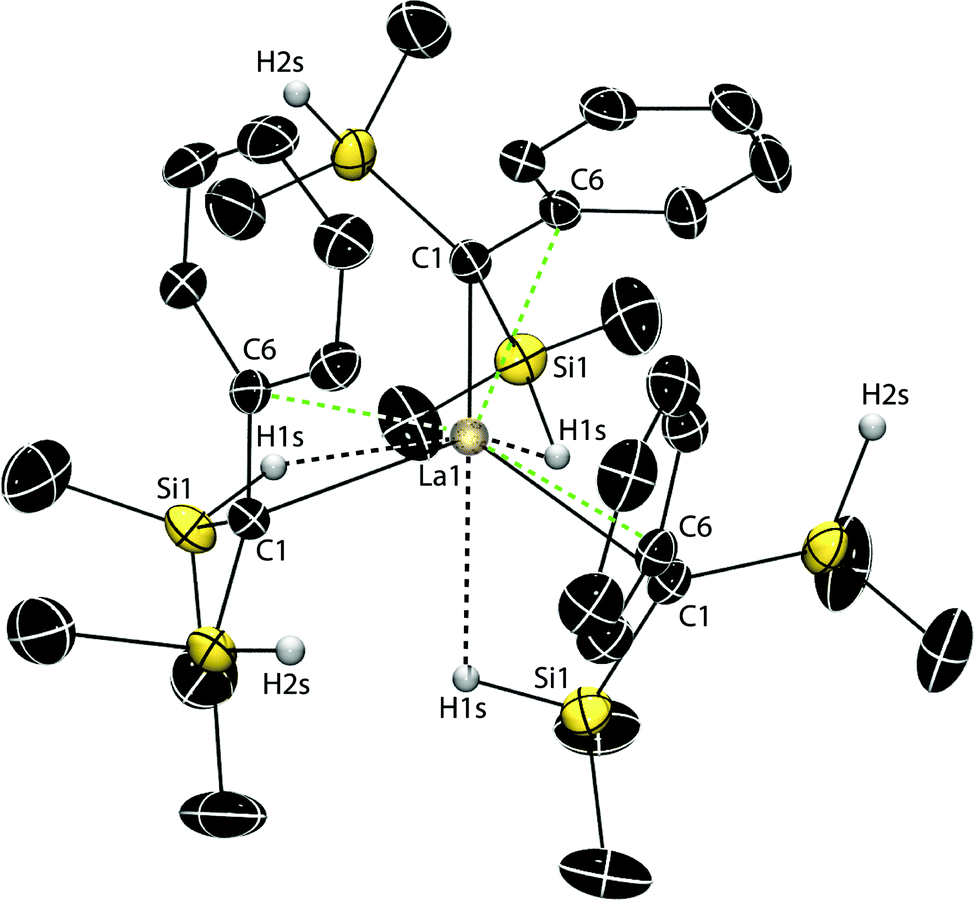
A single-crystal X-ray diffraction study revealed a polymeric structure for 2·TMEDA,‡ with each K cation interacting with two C(SiHMe2)2Ph groups (Fig. 1) through the H1s (2.82(4) Å), the C6 and C11 (from a phenyl group) of one ligand, and the C9 and C10 from a phenyl of the second. Notably, the K1–C1 distance (3.565(4) Å; i.e., to the presumed carbanionic center) is exceedingly long and outside expected bonding range. For comparison, the K–C distance (3.030(5) Å) is much shorter in dimeric {(Me2HSi)3CK(TMEDA)}2 than in 2·TMEDA.16 The SiH-free potassium alkyl {KC(SiMe3)2Ph}n (3.007(2) Å)8 and the compound {KC(SiMe2Ph)2(SiHMe2)}n (3.167(8) Å)20 also have polymeric structures with π-coordinated arenes. While the central, carbanionic carbon adopts distorted, nearly planar geometries in these three examples (Σangles = 358.8°, 357.4° and 356.5°),8,16,20 C1 in 2·TMEDA is perfectly planar (Σangles = 360.0(5)°).
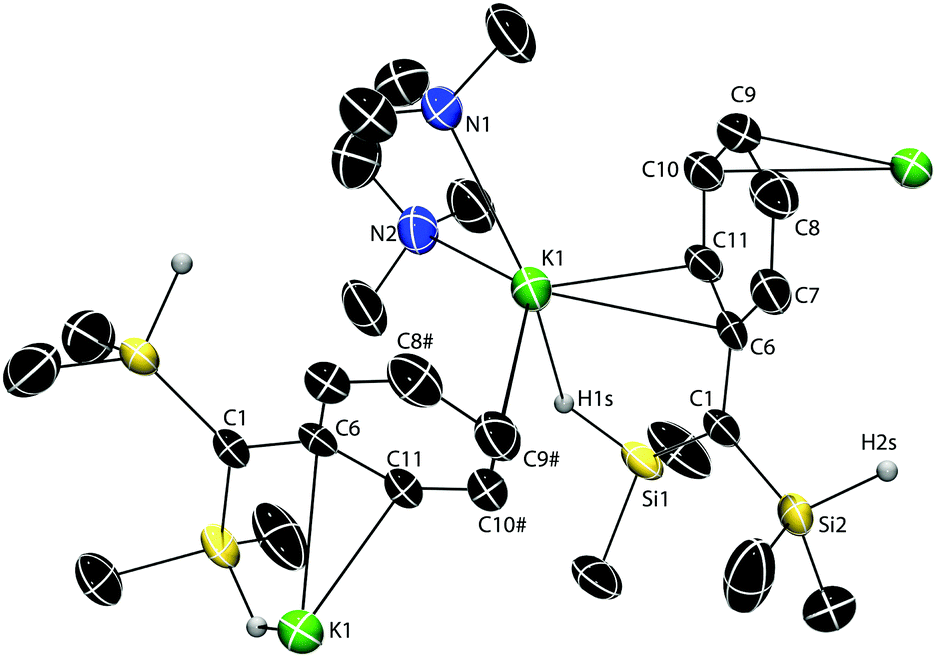 | ||
| Fig. 1 Thermal ellipsoid plot of Ph(Me2HSi)2CK(TMEDA) (2·TMEDA) at 50% probability. H1s and H2s were located objectively in the Fourier difference map and refined. H atoms bonded to C are not illustrated for clarity. Selected interatomic distances (Å): K1–C1, 3.565(4); K1–H1s, 2.82(4); K1–Si1, 3.844(2); K1–C6, 3.049(4); K1–C7, 3.389(5); K1–C8#, 3.359(5); K1–C9#, 3.085(5); K1–C10#, 3.038(4); K1–C11, 3.091(5); C1–C6, 1.446(6); C6–C7, 1.434(7); C7–C8, 1.376(7); C8–C9, 1.392(7); C9–C10, 1.383(7); C10–C11, 1.374(6); C11–C6, 1.422(6). Selected interatomic angles (°): K1–H1s–Si1, 124(2); C6–C1–Si2, 119.8(3); Si2–C1–Si1, 121.6(2); Si1–C1–C6, 118.6(3). | ||
The formation of Bn2SiMe2 in these reactions implies nucleophilic attack by KBn on a SiHMe2 group and suggests an alternative route to the desired KC(SiHMe2)2Ph (2) via Si–C cleavage. A related Si–Si bond cleavage provides MSi(SiMe3)3 from Si(SiMe3)4 and LiMe or KOtBu.21 This idea was tested by the reaction of (Me2HSi)3CPh and KOtBu to give the desired KC(SiHMe2)2Ph (2) in excellent yield (eqn (3)).
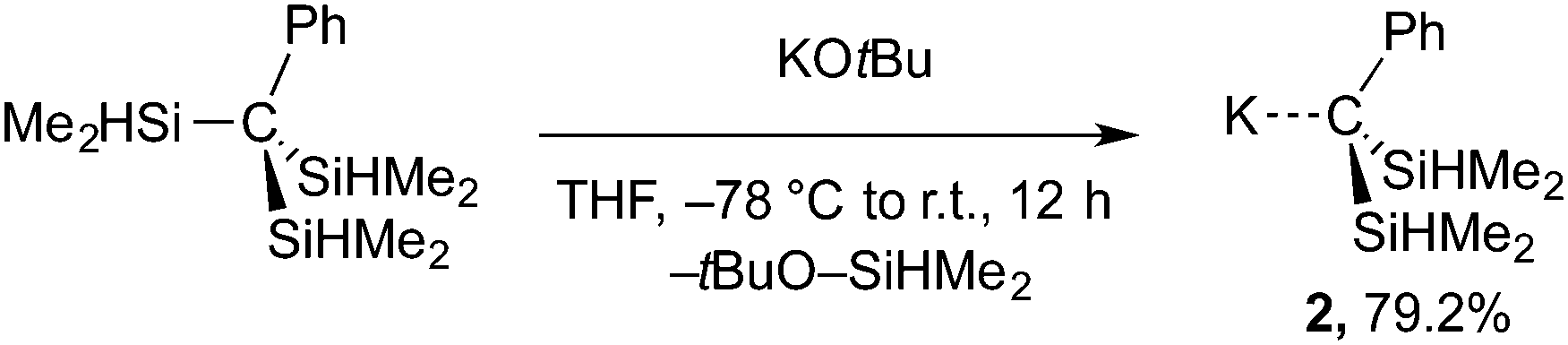 | (3) |
Reactions of three equiv. of 2 or 2·TMEDA and LaI3(THF)4, CeI3(THF)4, PrI3(THF)3, or NdI3(THF)3 provide Ln{C(SiHMe2)2Ph}3 (Ln = La (3), Ce (4), Pr (5), Nd (6)) in excellent yields (eqn (4)).
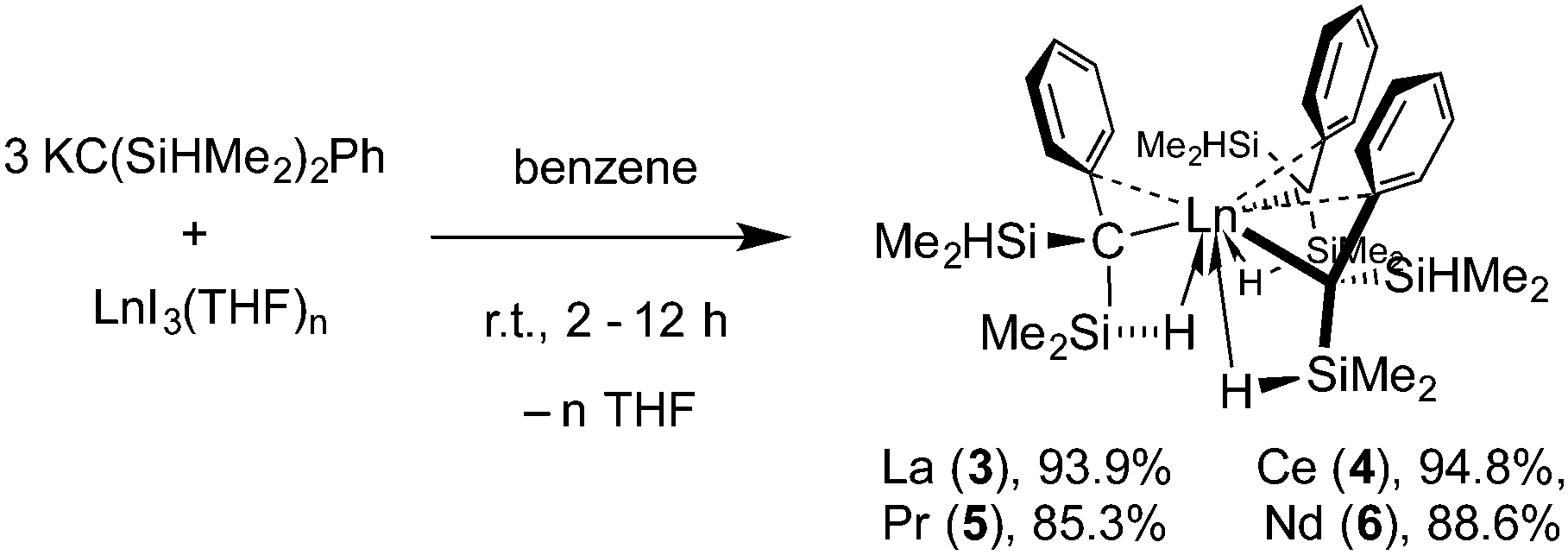 | (4) |
The series of compounds provide pale yellow, orange, yellow and green crystalline materials, respectively. IR spectra for 3, 5, and 6 (KBr) each contained a sharp, higher energy νSiH band (2109 ± 5 cm−1) assigned to non-bridging SiH and a broad, lower energy band (1866 ± 6 cm−1) attributed to a νSiH of the Ln↼H–Si. In contrast, the cerium compound 4 showed only one νSiH band, which appeared at 2115 cm−1. The solution IR spectra similarly showed two SiH absorbances for 3, 5 and 6. Although two νSiH bands were obtained for 4 in solution, the bands were low intensity and a number of the IR spectra were dominated by HC(SiHMe2)2Ph. We attributed these observations to labile secondary Ln↼H–Si interactions present both in solution and solid state.
The 1H NMR spectrum of diamagnetic 3 revealed signals at 4.24, 0.43, and 0.32 ppm attributed to equivalent SiHMe2 groups with diastereotopic methyl moieties. A doublet resonance at 5.84 ppm, assigned to an ortho-C6H5, appeared upfield compared to its chemical shift in the alkane starting material (6.97 ppm), suggesting a multihapto benzyl-Ln coordination. Evidence for La↼H–Si interactions were provided by the 1JSiH of 144 Hz. The equivalence of the SiHMe2 in the room temperature NMR spectrum contrasts the two types of SiH groups observed in the IR spectra, suggesting fluxional process(es). A 1H NMR spectrum collected at −73 °C in toluene-d8 revealed that two SiHMe2 groups were inequivalent: signals at 4.67 (1JSiH ∼ 180 Hz) and 3.82 (1JSiH ∼ 120 Hz) ppm were assigned to nonbridging SiH and bridging Ln↼H–Si moieties, respectively. These resonances correlated in a COSY experiment to signals at 0.35 and 0.08 ppm (with the downfield SiH) and 0.77 and 0.67 ppm (with the upfield SiH) of the now inequivalent methyl groups. Notably, one of the ortho-C6H5, whose resonance appeared unusually upfield at 4.15 ppm, was even more shielded than the nonbridging SiH. Moreover, all five H in the C6H5 were inequivalent. Thus, the alkyl ligands are equivalent in the low temperature solution-phase structure, with each ligand containing one La↼H–Si and a π-coordinated aryl group.
Single crystal X-ray diffraction reveals that the molecular structure of 3 (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) contains three, crystallographically related C(SiHMe2)2Ph ligands, each of which interacts with the lanthanum center through the central carbon (C1), through a benzylic-type coordination of the C6, and also through one La↼H–Si (Fig. 2).§ The three ligands are arranged in a trigonal geometry around the lanthanum center (ΣC1–La1–C1 = 357.15(6)°). This structure is consistent with the low temperature NMR and IR spectroscopic data. A few of the notable structural features include the sharp La1–C1–Si1 angle (93.0(1)°), short La1⋯Si1 and La1⋯H1s distances (3.3141(9) and 2.69(4) Å), and an unusually long La1–C1 distance (2.674(3) Å). The corresponding La–C distances in six-coordinate tris(benzyl)lanthanum compounds, e.g., La(CH2Ph)3(THF)3 (2.648(2) Å),5 are shorter, and the distance in La(CH(SiMe3)2)3 (2.515(9) Å) is much shorter.12 These trends extend to the comparisons of structures of 4–6 to the analogous benzyllanthanide species. Moreover, the close contacts in the series (i.e., Ln–C, Ln⋯Si, and Ln⋯H) follow the expected trend based on ionic radius (La > Ce > Pr > Nd).
) contains three, crystallographically related C(SiHMe2)2Ph ligands, each of which interacts with the lanthanum center through the central carbon (C1), through a benzylic-type coordination of the C6, and also through one La↼H–Si (Fig. 2).§ The three ligands are arranged in a trigonal geometry around the lanthanum center (ΣC1–La1–C1 = 357.15(6)°). This structure is consistent with the low temperature NMR and IR spectroscopic data. A few of the notable structural features include the sharp La1–C1–Si1 angle (93.0(1)°), short La1⋯Si1 and La1⋯H1s distances (3.3141(9) and 2.69(4) Å), and an unusually long La1–C1 distance (2.674(3) Å). The corresponding La–C distances in six-coordinate tris(benzyl)lanthanum compounds, e.g., La(CH2Ph)3(THF)3 (2.648(2) Å),5 are shorter, and the distance in La(CH(SiMe3)2)3 (2.515(9) Å) is much shorter.12 These trends extend to the comparisons of structures of 4–6 to the analogous benzyllanthanide species. Moreover, the close contacts in the series (i.e., Ln–C, Ln⋯Si, and Ln⋯H) follow the expected trend based on ionic radius (La > Ce > Pr > Nd).
 | ||
| Fig. 2 Thermal ellipsoid plot of La(C(SiHMe2)2Ph)3 (3). La1 is located on a crystallographic 3-fold axis. H atoms bonded to Si are located in the Fourier difference map, their positions are refined and are illustrated. All other H atoms and a disordered pentane molecule (0.5) are not shown for clarity. Short La–C (green) and Ln↼H–Si (black) distances are highlighted with dashed lines. Selected interatomic distances (Å): La1–C1, 2.674(3); La1–C6, 2.822(2); La1–Si1, 3.3141(9); La1–H1s, 2.69(4); C1–Si1, 1.821(3); Si1–H1s, 1.37(4); C1–Si2, 1.853(3); Si2–H2s, 1.45(4); C1–C6, 1.483(4); C6–C7, 1.415(5); C6–C11, 1.409(4). Selected interatomic angles (°): C1–La1–C1, 119.05(2); La1–C1–C6, 80.0(1); La1–C1–Si1, 93.0(1); La1–C1–Si2, 128.3(1). | ||
Interestingly, isomorphous cerium 4, praseodymium 5 and neodymium 6 compounds' structures (P21/c) are inequivalent with that of La 3. The two molecules in the unit cells for 4, 5 and 6 have inequivalent configurations, with one of the molecules containing only two Ln↼H–Si bridging moieties (Fig. 3).¶ The Ce–C distances for the five ligands that contain bridging Ce↼H–Si interactions average 2.65 ± 0.02 Å, whereas the η2-benzyl-only ligand (Ce2–C56, 2.587(3) Å) distance is shorter. This distinction is also apparent in compounds 5 (Pr–Cave, 2.63 ± 0.02; Pr2–C56, 2.556(5) Å) and 6 (Nd–Cave, 2.61 ± 0.02; Nd2–C56, 2.541(4) Å).
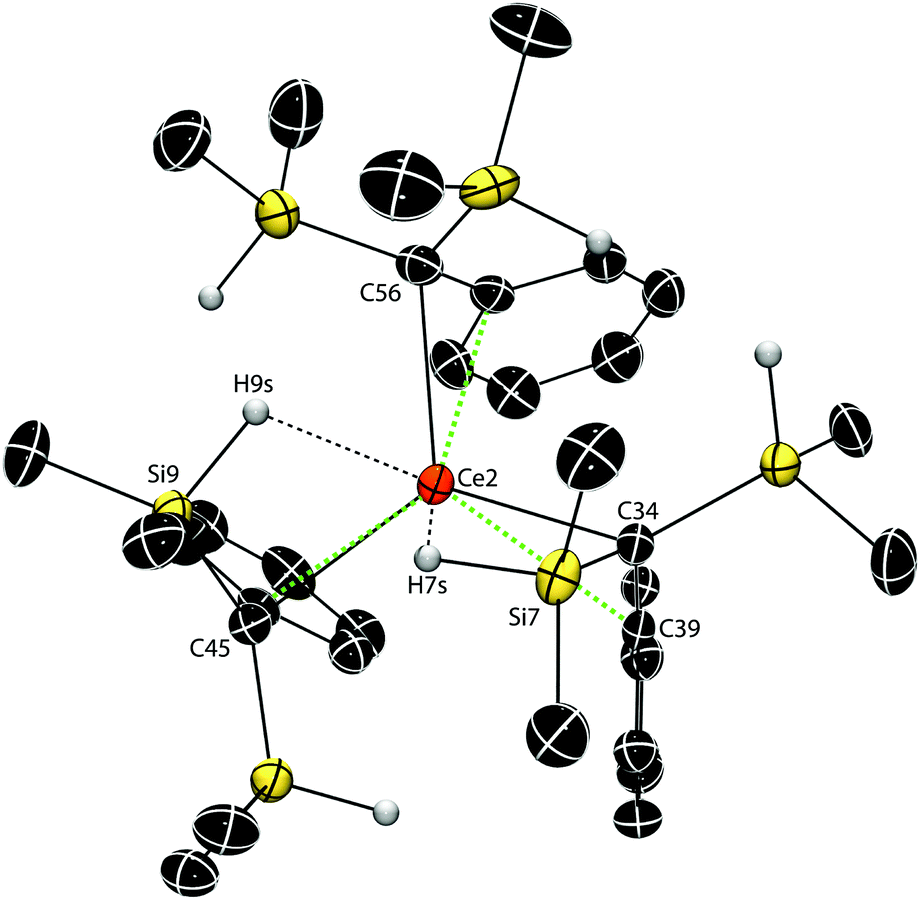 | ||
| Fig. 3 Thermal ellipsoid plot of one of two crystallographically distinct molecules of Ce(C(SiHMe2)2Ph)3 (4). H atoms bonded to Si are located in the Fourier difference map, their positions are refined and are illustrated. All other H atoms are not shown for clarity. Short Ce–C (green) and Ln↼H–Si (black) distances are highlighted with dashed lines. Selected interatomic distances (Å): Ce2–C34, 2.613(3); Ce2–C45, 2.671(2); Ce2–C56, 2.587(3); Ce2–Si7, 3.1947(9); Ce2–H7s, 2.47(2); C34–Si7, 1.829(2); Si7–H7s, 1.48(3); Ce2–Si9, 3.2379(9); Ce2–H9s, 2.46(3); C45–Si9, 1.829(3); Si9–H9s, 1.48(3); Selected interatomic angles (°): C34–Ce2–C45, 118.57(9); Ce2–C34–C39, 83.8(2); Ce2–C34–Si7, 90.1(1). | ||
This work extends the idea that the β-SiH group supports large, coordinatively unsaturated rare earth centers in homoleptic, solvent-free compounds to include a new mixed benzyl dimethylsilyl ligand. Tris(alkyl) lanthanides Ln{C(SiHMe2)2Ph}3 are synthesized in good yields, and the secondary interactions involving β-SiH and aryl moieties are likely important to the facile isolation of these compounds. The structural parameters (e.g., Ln–C, Si–H, Si–C distances) for moieties involved in secondary Ln↼H–Si interactions are different than those with nonbridging SiH groups, and the Ln–C distances are also affected by the presence or lack of secondary Ln↼H–Si interactions. The ligand itself is synthesized by deprotonation of the new alkane HC(SiHMe2)2Ph with KBn, but Me2SiBn2 and other side products in reactions of HC(SiHMe2)2Ph and KBn suggested a competing reaction involving nucleophilic attack on a Si center to cleave the C–Si bond. Therefore we developed an alternative route to KC(SiHMe2)2Ph by reacting PhC(SiHMe2)3 with KOtBu that affords the desired product in excellent yield. This straightforward two-step synthesis to homoleptic organolanthanides may allow their application in the preparation of heteroleptic lanthanide complexes and as precursors for new catalytic chemistry.
This research was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under Contract No. DE-AC02-07CH11358.
References
- M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194–6259 CrossRef CAS PubMed.
- A. M. Kawaoka, M. R. Douglass and T. J. Marks, Organometallics, 2003, 22, 4630–4632 CrossRef CAS; H. F. Yuen and T. J. Marks, Organometallics, 2008, 27, 155–158 CrossRef; H. F. Yuen and T. J. Marks, Organometallics, 2009, 28, 2423–2440 CrossRef; M. P. Conley, G. Lapadula, K. Sanders, D. Gajan, A. Lesage, I. del Rosal, L. Maron, W. W. Lukens, C. Copéret and R. A. Andersen, J. Am. Chem. Soc., 2016, 138, 3831–3843 CrossRef PubMed; M. Zimmermann, K. W. Törnroos and R. Anwander, Organometallics, 2006, 25, 3593–3598 CrossRef; H. Martin Dietrich, G. Raudaschl-Sieber and R. Anwander, Angew. Chem., Int. Ed., 2005, 44, 5303–5306 CrossRef PubMed; R. Taube, S. Maiwald and J. Sieler, J. Organomet. Chem., 2001, 621, 327–336 CrossRef; S. R. Daly, D. Y. Kim, Y. Yang, J. R. Abelson and G. S. Girolami, J. Am. Chem. Soc., 2010, 132, 2106–2107 CrossRef PubMed.
- H. Schumann, J. Muller, N. Bruncks, H. Lauke, J. Pickardt, H. Schwarz and K. Eckart, Organometallics, 1984, 3, 69–74 CrossRef CAS.
- A. J. Wooles, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 39, 500–510 RSC.
- S. Bambirra, A. Meetsma and B. Hessen, Organometallics, 2006, 25, 3454–3462 CrossRef CAS.
- A. C. Behrle and J. A. R. Schmidt, Organometallics, 2011, 30, 3915–3918 CrossRef CAS.
- S. Harder, Organometallics, 2005, 24, 373–379 CrossRef CAS.
- F. Feil and S. Harder, Organometallics, 2000, 19, 5010–5015 CrossRef CAS.
- P. L. Watson, J. Am. Chem. Soc., 1982, 104, 337–339 CrossRef CAS.
- H. Van der Heijden, C. J. Schaverien and A. G. Orpen, Organometallics, 1989, 8, 255–258 CrossRef CAS.
- P. L. Watson, J. Chem. Soc., Chem. Commun., 1983, 276–277 RSC.
- P. B. Hitchcock, M. F. Lappert, R. G. Smith, R. A. Bartlett and P. P. Power, J. Chem. Soc., Chem. Commun., 1988, 1007–1009 RSC.
- A. G. Avent, C. F. Caro, P. B. Hitchcock, M. F. Lappert, Z. N. Li and X. H. Wei, J. Chem. Soc., Dalton Trans., 2004, 1567–1577 RSC.
- W. A. Herrmann, J. Eppinger, M. Spiegler, O. Runte and R. Anwander, Organometallics, 1997, 16, 1813–1815 CrossRef CAS; J. William, S. Rees, O. Just, H. Schumann and R. Weimann, Angew. Chem., Int. Ed. Engl., 1996, 35, 419–422 CrossRef.
- K. Yan, A. V. Pawlikowski, C. Ebert and A. D. Sadow, Chem. Commun., 2009, 656–658 RSC; K. Yan, B. M. Upton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2009, 131, 15110–15111 CrossRef CAS PubMed; A. Pindwal, A. Ellern and A. D. Sadow, Organometallics, 2016, 35, 1674–1683 CrossRef.
- K. Yan, G. Schoendorff, B. M. Upton, A. Ellern, T. L. Windus and A. D. Sadow, Organometallics, 2013, 32, 1300–1316 CrossRef CAS.
- A. Pindwal, S. Patnaik, W. C. Everett, A. Ellern, T. L. Windus and A. D. Sadow, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201610263.
- C. Eaborn, P. B. Hitchcock and P. D. Lickiss, J. Organomet. Chem., 1983, 252, 281–288 CrossRef CAS.
- E. J. Hawrelak, F. T. Ladipo, D. Sata and J. Braddock-Wilking, Organometallics, 1999, 18, 1804–1807 CrossRef CAS.
- A. Asadi, A. G. Avent, M. P. Coles, C. Eaborn, P. B. Hitchcock and J. D. Smith, J. Organomet. Chem., 2004, 689, 1238–1248 CrossRef CAS.
- C. Marschner, Eur. J. Inorg. Chem., 1998, 221–226 CrossRef CAS; G. Gutekunst and A. G. Brook, J. Organomet. Chem., 1982, 225, 1–3 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization for compounds 1, 2, 2·TMEDA, 3–6. CCDC 1517682–1517686. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc09304c |
‡ X-ray data for 2·TMEDA (CCDC 1517682): C17H35KN2Si2; FW 362.75; monoclinic; a: 8.7354(6), b: 23.003(1), c: 11.6433(7), β: 100.207(4), volume: 2302.5(3); P121/c1; Z = 4; temp. 173 K; reflections: collected, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 907; independent, 3284; Rint 0.1552; 2217 data I > 2σ(I): R1 0.0705, wR2 0.1845; Rall: R1 0.0996, wR2 0.2145. 907; independent, 3284; Rint 0.1552; 2217 data I > 2σ(I): R1 0.0705, wR2 0.1845; Rall: R1 0.0996, wR2 0.2145. |
| § X-ray data for 3 (CCDC: 1517683): C33H57LaSi6·(C5H12)1/2; FW 797.31; trigonal; a: 12.4380(6), c: 16.157(1), volume, 2164.7(3); P; Z = 2; temp. 173 K; reflections: collected, 23107; independent, 3715; Rint 0.0345; 3439 data I > 2σ(I): R1 0.0309, wR2 0.0828; Rall: R1 0.0351, wR2 0.0928. |
| ¶ X-ray data for 4 (CCDC: 1517684): C33H57CeSi6; FW 762.44; monoclinic; a: 22.324(2), b: 19.841(1), c: 19.921(1), β: 116.076(1), volume: 7925.5(9); P121/c1; Z = 8; temp. 173 K; reflections: collected, 79198; independent, 18762; Rint 0.0485; 14549 data I > 2σ(I): R1 0.0315, wR2 0.0698; Rall: R1 0.0515, wR2 0.0836; X-ray data for 5 (CCDC 1517686): C33H57PrSi6; FW 763.23; monoclinic; a: 22.3310(6), b: 19.8399(5), c: 19.8802(5), β: 116.047(1), volume: 7913.2(4); P121/c1; Z = 8; temp. 173 K; reflections: collected, 79681; independent, 15456; Rint 0.0653; 14828 data I > 2σ(I): R1 0.0695, wR2 0.1909; Rall: R1 0.0710, wR2 0.1931; X-ray data for 6 (CCDC: 1517685): C33H57NdSi6; monoclinic; a: 22.335(2), b: 19.843(2), c: 19.855(2), β: 116.017(1), volume: 7908(1); P121/c1; Z = 8; temp. 173 K; reflections: collected, 113017; independent, 20627; Rint 0.0711; 15075 data I > 2σ(I): R1 0.0346, wR2 0.0623; Rall: R1 0.0624, wR2 0.0768. |
| This journal is © The Royal Society of Chemistry 2017 |