DOI:
10.1039/C6CC08970D
(Communication)
Chem. Commun., 2017,
53, 697-700
Sumanene derivatives functionalized at the internal carbon†
Received
10th November 2016
, Accepted 25th November 2016
First published on 25th November 2016
Abstract
The first sumanene derivatives functionalized at the convex face of the internal carbon were reported. The structure of racemic hydroxysumanenone was confirmed by X-ray crystal analysis. The obtained structure was composed of the single enantiomer.
Bowl-shaped aromatic hydrocarbons, known as buckybowls, are attractive molecules due to their unique aromatic bowl shape and their chemical and physical properties. Since they correspond to the partial structure of fullerenes, the similarities or differences in their properties compared with the latter are of great interest. In fullerenes' chemistry, a variety of addition reactions such as Bingel–Hirsch and Prato reactions have been developed for the derivatization from the pristine fullerenes (Scheme 1A).1–3 Since fullerenes do not have a peripheral edge, any addition reaction occurs on the internal carbons. In contrast, most derivatizations of buckybowls are substitution reactions at the peripheral carbons, while a limited number of addition reactions at the internal carbons have been reported in non-substituted pristine buckybowls4 such as corannulene,5 diindenochrysene,6 and circumtrindene7 without functional substituents (Scheme 1). Petrukhina et al. succeeded in generating a corannulenium cation through the stepwise carbene insertion process,5d which had been previously proposed by Scott (Scheme 1B).5e Scott also reported a nucleophilic addition by methylithium to the interior of diindenochrysene, followed by the methylation with methyl iodide (Scheme 1C).6 The Prato reaction was reported with a bigger buckybowl, circumtrindene (Scheme 1D).7a These 1,2-addition reactions of buckybowls, without a substituent effect, are similar to those of fullerenes, and they were the only method used to prepare buckybowl derivatives tethered to the convex internal position. On the other hand, there is no report on the functionalization of the pristine sumanene (1)8 at the internal carbon.
 |
| | Scheme 1 Examples of functionalization on the internal carbons of (A) C60, (B) corannulene, (C) diindenochrysene, and (D) circumtrindene. | |
However, in buckybowls, functionalization at the internal carbons using installed functional groups at the peripheral positions is also available in principle, in contrast to fullerenes without peripheral functional substituents. The existence of functional groups allows us to employ different types of reactions in preparing tethered buckybowl derivatives at the convex internal positions. In the course of our study on a sumanene derivative, hydroxysumanene,9 we found that bromination on hydroxysumanene with N-bromosuccinimide (NBS) followed by a nucleophilic substitution reaction produced substituted sumanene derivatives on the internal carbon of a sumanene framework (Scheme 2). This reaction demonstrated another derivatization approach of buckybowls, which prepares the buckybowl derivatives that are tethered to the convex internal position, unlike in fullerene chemistry. In this communication, we report that the syntheses of the first sumanene derivatives tethered by a hydroxy or methoxy group at the convex internal carbons, together with their crystal structures, chirality, and photophysical properties.
 |
| | Scheme 2 Functionalization on the internal carbon of hydroxysumanene 2. | |
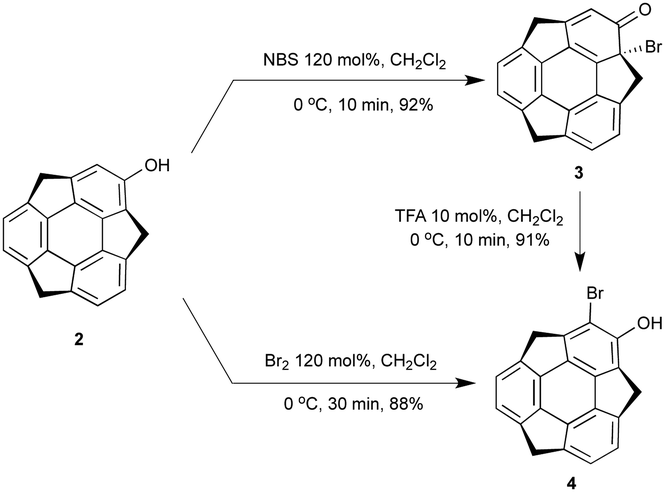
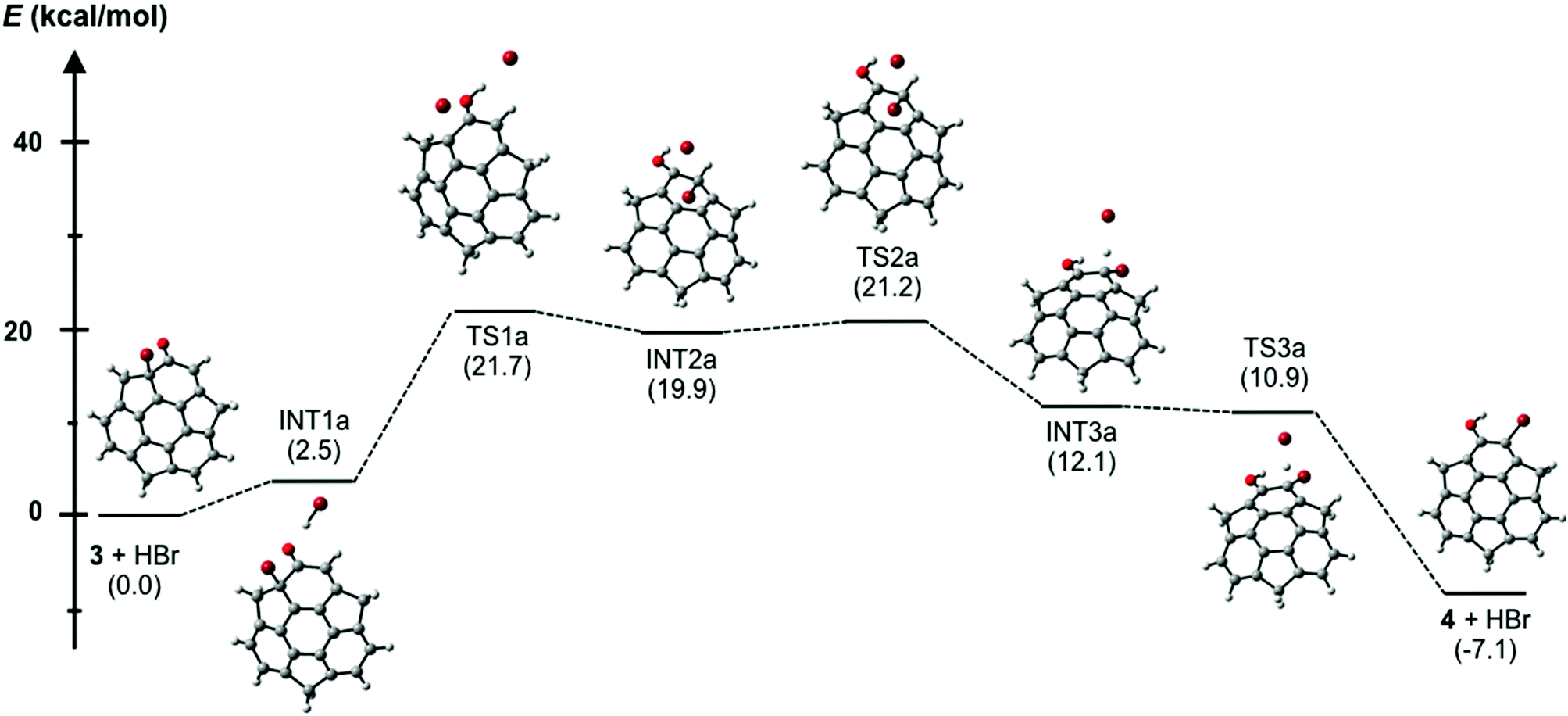
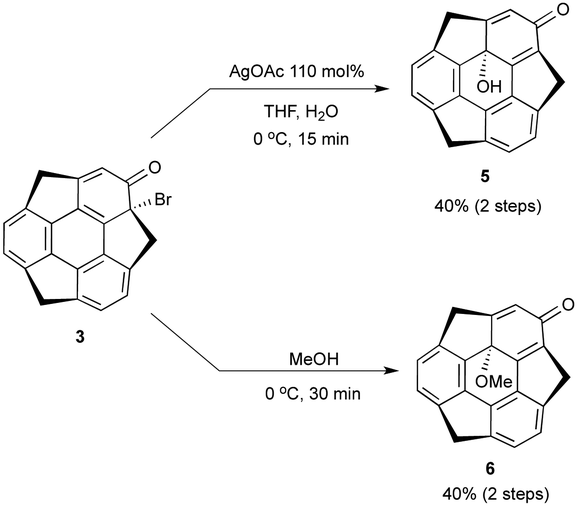
In the course of the derivatization study on hydroxysumanene (2), we found that bromosumanenone (3)10 possessing a dearomatized bromodienone structure was formed in 92% yield by bromination of 2 with NBS, while bromination of 2 with Br2 resulted in o-bromohydroxysumanene (4) in 88% yield (Scheme 3). This bromosumanenone 3 was unstable against acid, and was therefore converted into 4 immediately by addition of trifluoroacetic acid (TFA). A similar bromination reaction of phenolic compounds with NBS to produce bromodienone products was previously reported by Chow et al.11 They also reported that the migration of bromide and aromatization of bromodienone compounds to o-bromophenol were accelerated by acid. To understand the acid-assisted bromide migration from 3 to 4, we performed DFT calculations [ωB97XD/6-311G(d,p)//PCM/CH2Cl2] of the reaction from 3 with HBr to 4 (Scheme 4), which indicated that the migration of the bromonium cation (TS1a) is the rate determining step. We then further investigated the derivatization of 3. Its reaction with a stoichiometric amount of silver acetate in H2O/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded hydroxysumanenone (5) (Scheme 5) that possessed a hydroxy group at the internal carbon of the sumanene skeleton in 40% yield in two steps from 2. The structure of 5 was unambiguously determined by X-ray crystallographic analysis (vide infra). Similarly, methoxysumanenone (6) was also obtained in 40% yield in two steps from 2 by the solvolysis of 3 by methanol.
:1) afforded hydroxysumanenone (5) (Scheme 5) that possessed a hydroxy group at the internal carbon of the sumanene skeleton in 40% yield in two steps from 2. The structure of 5 was unambiguously determined by X-ray crystallographic analysis (vide infra). Similarly, methoxysumanenone (6) was also obtained in 40% yield in two steps from 2 by the solvolysis of 3 by methanol.
 |
| | Scheme 3 Bromination of 2 by Br2 or NBS. | |
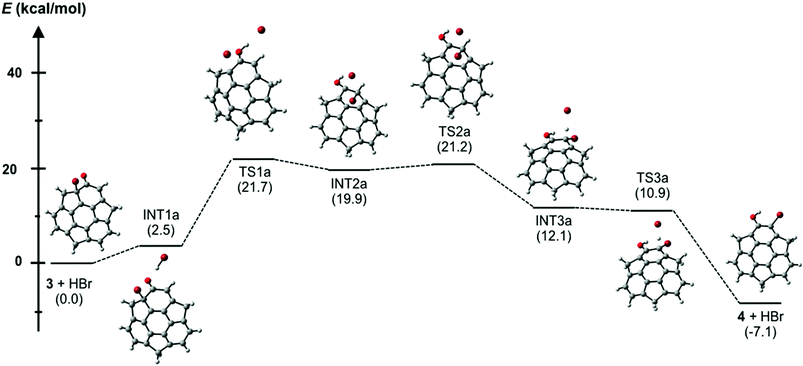 |
| | Scheme 4 Energy diagram of acid-mediated transformation from bromosumanenone 3 to bromohydroxysumanene 4 [ωB97XD/6-311G(d,p)//PCM/CH2Cl2]; the values in parentheses are relative free energies (298.15 K, 1.0 atm). | |
 |
| | Scheme 5 Synthesis of hydroxysumanenone 5 and methoxysumanenone 6 from 3. | |
5 and 6 are chiral compounds and do not racemize by bowl inversion owing to the existence of the convex hydroxy or methoxy group at the C16 stereogenic center unlike chiral buckybowls that only possess bowl chirality12,13 such as 2.9 When we analyzed the crystals of (±)-6 by X-ray diffraction, we found that a crystal is composed of only single enantiomers. We succeeded in separating the enantiomers of 5 and 6 using chiral supercritical fluid chromatography (SFC) (CHIRALPAX IA; co-solvent, CH2Cl2) (Fig. 1) and obtained each enantiomer with >99% ee [circular dichroism (CD) spectra of 5 and 6 in Fig. 2]. The X-ray analysis of the crystals of each enantiomer of 5 showed the same crystal structure as that of the chiral crystals of (±)-5. The crystal structure of (−)-5 is shown in Fig. 3. The space group of the crystal was P32 and the antipode (+)-5 gave the P31 system. This indicated that both of the crystals were composed of only one kind of enantiomer. Since the crystals of the racemic material contain only one enantiomer the system obviously results in spontaneous resolution upon crystallization. The hydroxy group was located on the C16 position on the convex face of the bowl. Due to the introduction of the substituent on this position, two 6-membered rings, the dienone ring (C1, C2, C17, C16, C14, and C15) and the bottom six-membered ring (C16, C17, C18, C19, C20, and C21), were dearomatized. Accordingly, the bowl depth that was measured between the plane composed of five atoms of the six-membered ring except C16 (C17, C18, C19, C20, C21; plane A) and three benzylic carbons (C3, C8, and C13) were 1.09, 0.92, and 0.77 Å, respectively (Fig. 3b). Meanwhile, the distances between the plane, which was parallel to plane A and included C16 (plane B), and the same three benzylic carbons were 1.43, 1.25, and 1.11 Å, respectively. These values are much greater than that of 1 (1.11 Å)13 and the nitrogen-doped sumanene derivative (1.30 Å),12 clearly indicating that the substitution on the internal carbon caused a significant distortion of the bowl structure. C3-symmetric helical structures were formed by strong OH⋯O type hydrogen bonds between the hydroxy and carbonyl groups of neighboring molecules with the same chirality (2.76 Å, Fig. 3c). These C3-symmetric helical structures stacked against each other to give a partially-overlapping unidirectional columnar structure along the c axis, which was totally different from that observed in pristine sumanene crystals.14 The C3-symmetric helical structures interacted with each other by CH⋯π interaction between benzylic CH and the aromatic ring moiety of 5 (Fig. 3d).
 |
| | Fig. 1 Chiral SFC chromatograms of (a) 5 (CH2Cl2 0.8 mL min−1, CO2 0.8 mL min−1) and (b) 6 (CH2Cl2 0.5 mL min−1, CO2 0.8 mL min−1) using the CHIRALPAX IA column. The UV and CD detector absorbed at 254 nm. | |
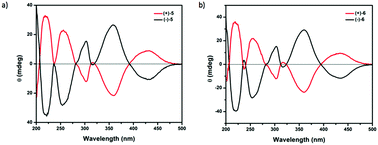 |
| | Fig. 2 Circular dichroism of (a) 5 and (b) 6 in CH3CN at 2.00 × 10−4 M. | |
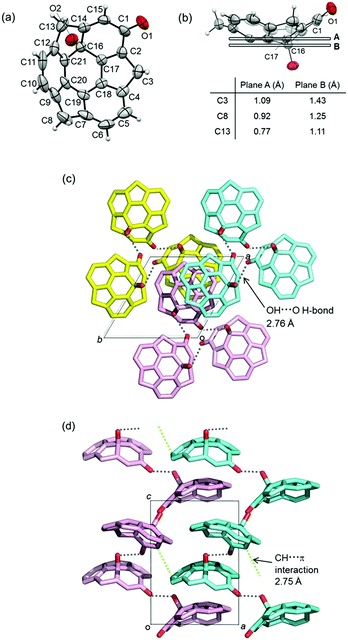 |
| | Fig. 3 Crystal structure of the single enantiomer of (−)-5. (a) ORTEP drawing of (−)-5 with 50% probability. (b) Bowl depth between two kinds of planes and three benzylic carbons C3, C8 and C13. (c) Packing structure viewed along the c axis. Each C3-symmetric helical structure is shown in yellow, pale purple and blue. (d) Packing structure viewed along the b axis. Gray, yellow, blue and pink are C atoms; red is O atoms. H atoms are omitted from (c) and (d) for clarity. | |
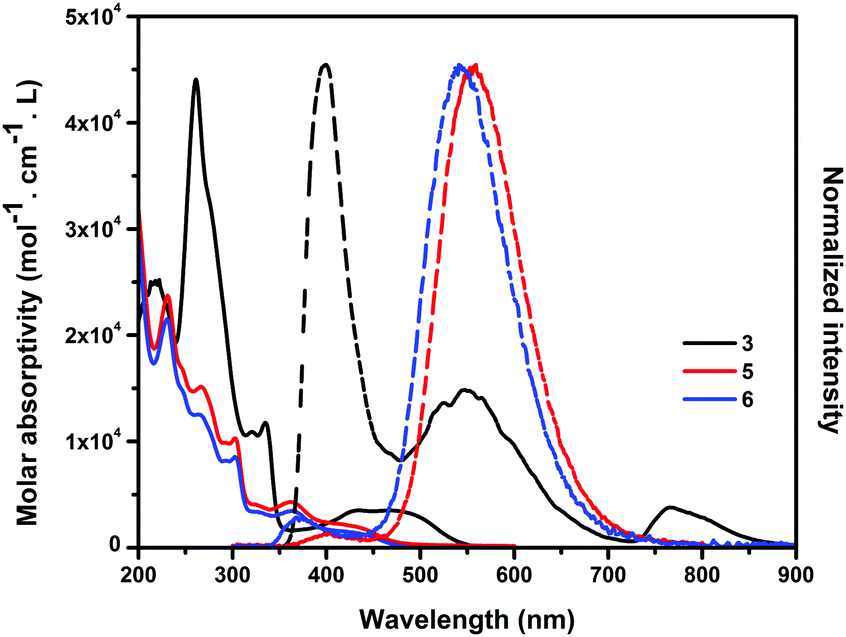
The UV-vis absorption and emission spectra of 3, 5 and 6 were measured in acetonitrile solution (Fig. 4). The absorption spectra of 5 and 6 showed similar shapes and patterns and were different from that of 3 due to the different substitution position as well as the different π-conjugated systems. 5 and 6 showed the absorption band at λmax = 230 nm (assigned to HOMO−2 to LUMO+1 transition), which was blue-shifted from that of 3 at λmax = 260 nm (assigned to HOMO−1 to LUMO+2). The observed absorption peaks of 5 and 6 at 445 nm corresponded to their HOMO to LUMO transition states. 3 showed an absorption band at 433 and 490 nm, assignable to the HOMO−1 to LUMO and the HOMO to LUMO transition states, respectively. The absorption edges of 5 and 6 (460 nm) were blue-shifted from that of 3 (547 nm), indicating a larger HOMO–LUMO gap than 3. Emission spectra of 3 excited at the absorption maximum (260 nm) were observed at 400, 552 and 762 nm. The emission maximum of 3 was approx. 150 nm blue-shifted than those of 5 (560 nm) and 6 (530 nm).
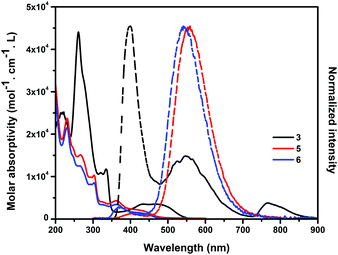 |
| | Fig. 4 Absorption (solid line) and emission (dashed line) spectra of 3, 5 and 6 in CH3CN. The emission spectra were excited at the absorption maxima. | |
In conclusion, we succeeded in the functionalization at the internal carbon of hydroxysumanene 2 and synthesizing first tethered sumanene derivatives at the convex internal position. Bromination of 2 with NBS gave bromosumanenone 3 and the following nucleophilic substitution reaction afforded hydroxysumanenone 5 or methoxysumanenone 6 possessing the hydroxy or methoxy group at the internal carbon of the sumanene skeleton. The enantiomers of 5 and 6 were separated by chiral SFC and the chiral crystals of 5 were obtained. The unidirectional helical packing structure and strong interaction between columns were observed via OH⋯O type hydrogen bonds between the hydroxy group on the internal position and the carbonyl groups of the adjacent column.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Area “π Space Figuration” from MEXT (No. 26102002), and JSPS KAKENHI (26288020). S. H. acknowledges ACT-C, JST, for the support. The authors appreciate the great help from Prof. Norimitu Tohnai with single crystal X-ray diffraction experiments.
Notes and references
- For examples of Bingel–Hirsch reaction, see
(a) G. E. Ball, G. A. Burley, L. Chaker, B. C. Hawkins, J. R. Williams, P. A. Keller and S. G. Pyne, J. Org. Chem., 2005, 70, 8572–8574 CrossRef CAS PubMed;
(b) Y. Nakamura, M. Suzuki, Y. Imai and J. Nishimura, Org. Lett., 2004, 6, 2797–2799 CrossRef CAS PubMed;
(c) M. Á. Herranz, C. T. Cox Jr. and L. Echegoyen, J. Org. Chem., 2003, 68, 5009–5012 CrossRef CAS PubMed;
(d) X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 1595–1596 RSC;
(e) C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- For examples of Prato reaction, see
(a) S. Filippone, M. I. Barroso, Á. M. Domonech, S. Osuma, M. Solà and N. Martín, Chem. – Eur. J., 2008, 14, 5198–5206 CrossRef CAS PubMed;
(b) M. Prato, Acc. Chem. Res., 1998, 31, 519–526 CrossRef CAS;
(c) S. R. Wilson and Q. Lu, J. Org. Chem., 1995, 60, 6496–6498 CrossRef CAS;
(d) M. Maggini and G. Scorrano, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- For other reactions with fullerenes, see
(a) N. Ikuma, K. Nakagawa, K. Kokubo and T. Oshima, Org. Biomol. Chem., 2016, 14, 7103–7108 RSC;
(b) W. T. Su, M. Watanabe, Y. J. Chang, P. T. Chou, A. Ghosh and T. J. Chow, Tetrahedron Lett., 2015, 56, 1092–1095 CrossRef CAS;
(c) Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657–4708 RSC;
(d) R. Kessinger, J. Crassous, A. Herrmann, M. Rüttimann, L. Echegoyen and F. Diederich, Angew. Chem., Int. Ed., 1998, 37, 1919–1922 CrossRef CAS;
(e) C. Thilgen, A. Herrmann and F. Diederich, Angew. Chem., 1997, 109, 2362–2374 (
Angew. Chem., Int. Ed. Engl.
, 1997
, 36
, 2268–2280
) CrossRef.
-
(a) L. T. Scott, Chem. Soc. Rev., 2015, 44, 6464 RSC;
(b) L. T. Scott, H. E. Bronstein, D. V. Preda, R. B. M. Ansems, M. S. Bratcher and S. Hagen, Pure Appl. Chem., 1999, 71, 209–219 CrossRef CAS.
-
(a) C. Dubceac, A. S. Filatov, A. V. Zabula and M. A. Petrukhina, Cryst. Growth Des., 2015, 15, 778–785 CrossRef CAS;
(b) C. Dubceac, A. V. Zabula, A. S. Filatov, F. Rossi, P. Zanello and M. A. Petrukhina, J. Phys. Org. Chem., 2012, 25, 553–558 CrossRef CAS;
(c) M. Yanney, F. R. Fronczek and A. Sygula, Org. Lett., 2012, 14, 4942–4945 CrossRef CAS PubMed;
(d) A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2011, 50, 2971–2974 CrossRef CAS PubMed;
(e) D. V. Preda and L. T. Scott, Tetrahedron Lett., 2000, 41, 9633–9637 CrossRef CAS.
- H. E. Bronstein and L. T. Scott, J. Org. Chem., 2008, 73, 88–93 CrossRef CAS PubMed.
-
(a) H. Y. Cho, R. B. M. Ansems and L. T. Scott, Beilstein J. Org. Chem., 2014, 10, 956–968 CrossRef PubMed;
(b) J. A. Steckel and K. D. Jordan, J. Phys. Chem. A, 2002, 106, 2572–2579 CrossRef CAS.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- N. Ngamsomprasert, G. Panda, S. Higashibayashi and H. Sakurai, J. Org. Chem., 2016, 81, 11978–11981 CrossRef CAS PubMed.
- For the determination of the structures of 3 and 4, see the ESI†.
- Y. L. Chow, D. C. Zhao and C. I. Johansson, Can. J. Chem., 1988, 66, 2556–2564 CrossRef CAS.
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed.
-
(a) S. Higashibayashi, R. Tsuruoka, Y. Soujanya, U. Purushotham, G. N. Sastry, S. Seki, T. Ishikawa, S. Toyota and H. Sakurai, Bull. Chem. Soc. Jpn., 2012, 85, 450–467 CrossRef CAS;
(b) R. Tsuruoka, S. Higashibayashi, T. Ishikawa, S. Toyota and H. Sakurai, Chem. Lett., 2010, 39, 646–647 CrossRef CAS;
(c) S. Higashibayashi and H. Sakurai, J. Am. Chem. Soc., 2008, 130, 8592–8593 CrossRef CAS PubMed.
-
(a) S. Mebs, M. Weber, P. Luger, B. M. Schmidt, H. Sakurai, S. Higashibayashi, S. Onogi and D. Lentz, Org. Biomol. Chem., 2012, 10, 2218–2222 RSC;
(b) H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, J. Am. Chem. Soc., 2005, 127, 11580–11581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1512434 and 1512435. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08970d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence