
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic analysis of copper transfer from a chaperone to its target protein mediated by complex formation†
Kristine L.
Kay
a,
Liang
Zhou
a,
Leonardo
Tenori
b,
Justin M.
Bradley
a,
Chloe
Singleton
a,
Margaret A.
Kihlken
a,
Simone
Ciofi-Baffoni
b and
Nick E.
Le Brun
 *a
*a
aCentre for Molecular and Structural Biochemistry, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: n.le-brun@uea.ac.uk
bMagnetic Resonance Center CERM and Department of Chemistry, University of Florence, Via Luigi Sacconi 6, 50019, Sesto Fiorentino, Florence, Italy
First published on 5th January 2017
Abstract
Chaperone proteins that traffic copper around the cell minimise its toxicity by maintaining it in a tightly bound form. The transfer of copper from chaperones to target proteins is promoted by complex formation, but the kinetic characteristics of transfer have yet to be demonstrated for any chaperone-target protein pair. Here we report studies of copper transfer between the Atx1-type chaperone CopZ from Bacillus subtilis and the soluble domains of its cognate P-type ATPase transporter, CopAab. Transfer of copper from CopZ to CopAab was found to occur rapidly, with a rate constant at 25 °C of ∼267 s−1, many orders of magnitude higher than that for Cu(I) dissociation from CopZ in the absence of CopAab. The data demonstrate that complex formation between CopZ and CopAab, evidence for which is provided by NMR and electrospray ionisation mass spectrometry, dramatically enhances the rate of Cu(I) dissociation from CopZ.
Copper is both essential and potentially toxic to cells. To counter the latter, organisms have evolved a complex network of pathways through which copper, as Cu(I), is trafficked into, within and out of cells.1,2 Attention has focussed not only on identifying the proteins involved in such pathways, but also on the mechanisms by which Cu(I) is passed on from one protein to the next. It is generally accepted that, to minimise toxicity, such mechanisms must avoid the release of Cu(I) into solution. The most commonly found Cu(I)-trafficking pathways involve Atx1-like chaperones and their cognate Cu(I)-transporting P-type ATPases, which are found in many bacteria, yeast and in higher eukaryotes, including humans.2 Copper transfer from Atx1-like chaperones to their partner proteins has been demonstrated both in vivo and in vitro. A facile exchange mechanism was proposed for the transfer of Cu(I) from the chaperone to the N-terminal domain of the transporter,3 in which Cu(I) bound to the chaperone in a digonal or trigonal manner depending on sample conditions3,4 forms three-coordinate intermediates, with Cys residues from the transporter N-terminal domain supplying additional ligands. In this way, Cu(I) bridges the two proteins and the intermediate can be resolved in either direction.
Typically, the presence of cargo metal ions dramatically enhances the interaction between the chaperone and partner proteins.5,6 In addition, it has been shown in several systems that specific protein–protein molecular recognition via electrostatically complementary surfaces facilitates an interaction between the proteins7–9 and hence a mechanism for rapid exchange. From a thermodynamic perspective, it has been shown that Cu(I) is transferred along cellular copper trafficking pathways from chaperones to partner proteins exploiting gradients of copper-binding affinity.10 In the case of Cu(I) transfer from Atx1-like chaperones to the N-terminal domains of transporters, this gradient is able to move copper from the chaperone to the transporter, though in a few cases the gradient has been shown to be very shallow.11
In the cytoplasm of Bacillus subtilis, the Atx1-like chaperone CopZ delivers Cu(I) to the P-type ATPase CopA for extrusion.12 Structural and Cu(I)-binding properties of CopZ and the two N-terminal soluble domains of CopA (CopAab) have been well characterised.13–21 CopZ undergoes Cu(I)-mediated dimerization upon initial binding of Cu(I), with a stability constant of β2 ∼ 1023 M−2, and an estimated stability constant, K, for the initial Cu–CopZ complex of 1017–1018 M−1 (ref. 20). CopAab remains monomeric at low Cu(I) levels (<1 Cu/CopAab) but also binds Cu(I) with very high affinity (K = ∼1017 for the initial binding event18). Specific interaction between CopZ and CopAab was demonstrated by bacterial two-hybrid experiments12 and NMR studies revealed the formation of a weak complex between CopZ and the second soluble domain of CopA (CopAb) that was mediated by Cu(I), probably through a three-coordinate intermediate.22 Despite the wealth of thermodynamic and structural information about copper chaperones, there is little kinetic information about copper transfer available and the acceleration of Cu(I)-transfer driven by protein–protein interactions has not been demonstrated.
To facilitate the measurement of Cu(I) transfer between CopZ and CopAab, a C-terminally His6-tagged form of CopZ was generated with Cu(I)-binding properties indistinguishable from the native protein (Fig. S1, ESI†). Transfer experiments at a loading of 0.5 Cu(I) per protein were conducted in both directions, i.e., beginning with Cu(I)–CopZ and adding apo-CopAab and vice versa. The proteins were separated using Ni(II)-affinity chromatography and analysis of fractions for protein (by colorimetric determination and LC-MS) and Cu(I) revealed that Cu(I) was only present in protein-containing fractions and that it was equally distributed between the proteins, see Fig. 1 and Fig. S2 (ESI†). Similar transfer experiments were performed for 1 Cu(I)/CopZ and apo-CopAab (and vice versa), which also demonstrated transfer in both directions and an approximately equal distribution of Cu(I) between the proteins (0.6 ± 0.1 Cu(I)/CopAab), consistent with the similar Cu(I) affinities reported previously for CopZ and CopAab.14–21,23In vivo, CopAab is part of the much larger membrane-associated CopA transporter, and interactions between the two soluble domains and the membrane spanning segments24 could influence the Cu(I) affinity of the soluble domains, potentially enhancing the thermodynamic gradient for transfer of Cu(I) to the exporter. However, previous in vivo studies of a copZ deletion strain of B. subtilis revealed that it contained significantly less (∼3-fold) copper than the wild-type strain,12 suggesting that CopZ may act as a cellular copper store. Thus, CopZ is able to retain a significant proportion of Cu(I) in competition with CopAab, suggesting that B. subtilis requires copper in the cytoplasm; for example, for use during sporulation (the B. subtilis spore coat protein CotA is a copper-containing laccase).25
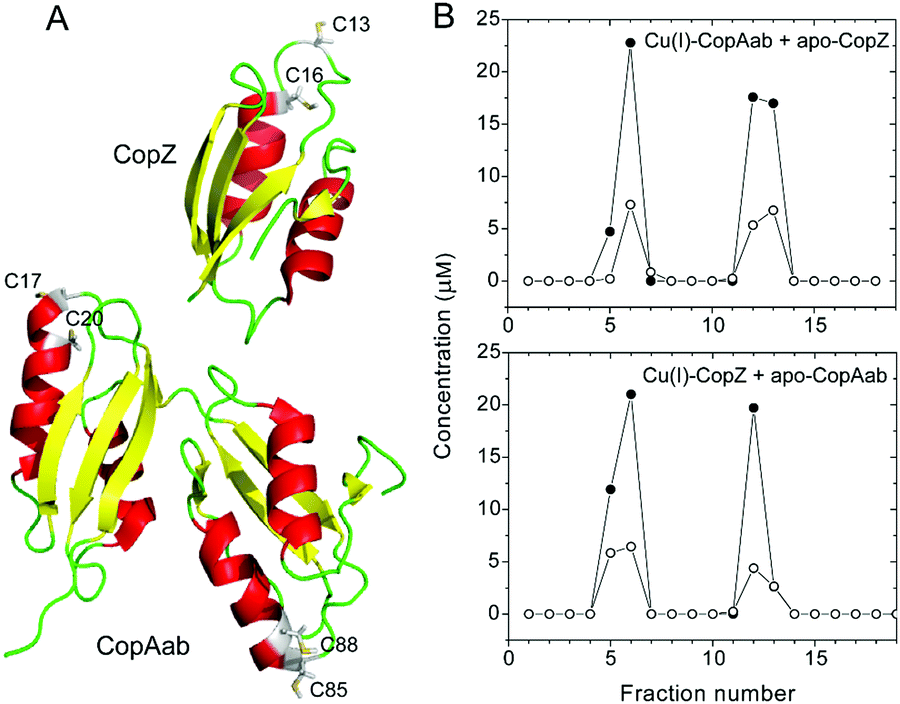 | ||
| Fig. 1 Determination of Cu(I)-transfer between CopZ and CopAab. (A) Ribbon diagrams depicting solution structures of CopZ and CopAab in their apo-form. The coordinating cysteines in the copper binding site are labelled and shown in grey, α-helices are shown in red, β-sheets shown in yellow, flexible linkers are shown in green. (B) Plot of protein (filled circles) and Cu(I) concentrations (open circles) determined for fractions collected from a Ni(II)-affinity column, where fractions 1–8 were eluted with buffer A and fractions 9–18 were eluted with 20 mM Tris-HCl, 0.5 M NaCl, 500 mM imidazole, pH 7.5. Top panel: Column was loaded with CopAab (170 μM) containing 0.5 Cu(I) per protein and apo-(His)6-CopZ (170 μM). Bottom panel: Column was loaded with (His)6-CopZ (170 μM) containing 0.5 Cu(I) per protein and apo-CopAab (170 μM). | ||
The stability constant for the Cu(I)–CopZ complex represents the ratio of dissociation and association rate constants. Because the stability constant is very high (∼1017 M−1) and the association rate constant is limited by the rate of diffusion to ∼109 M−1 s−1, the dissociation rate constant must be extremely low (predicted to be ∼10−8 s−1). Attempts to measure this by monitoring transfer of Cu(I) between proteins separated by semi-permeable membranes were hampered by the instability of CopZ in solution over long time periods, but absorbance measurements at 25 °C over 46 h revealed only small changes in absorbance (Fig. S3, ESI†), giving an upper limit estimate of the dissociation rate constant of ∼6 × 10−5 s−1. Thus, the data are consistent with an extremely low rate of dissociation of Cu(I) from the Cu(I)–CopZ complex, which represents a kinetic barrier to the transfer of Cu(I) to target proteins.
Kinetic information about the transfer of a single Cu(I) between chaperones and target proteins has not so far been reported, in part due to the difficulties of distinguishing between Cu(I) ions in very similar environments (associated with chaperone and target proteins). While binding of Cu(I) to both CopZ and CopAab gives rise to Cys thiolate-to-Cu(I) charge transfer transitions in the near UV,14,17 the intensity properties of these bands on a per Cu(I) basis are very different, with initial binding of Cu(I) to CopZ significantly more intense (see Fig. S4, ESI†). This differential response provides a means to monitor both the rate and direction of Cu(I) transfer between CopZ and CopAab. Thus, kinetics of Cu(I) transfer between CopZ and CopAab were followed by measuring absorbance changes at 265 nm by stopped-flow absorbance. While no changes were observed on mixing CopZ and CopAab in the absence of Cu(I) (Fig. S5, ESI†), they were upon mixing 0.5 Cu/CopZ with apo-CopAab at a range of concentrations at 25 °C, see Fig. 2A and Fig. S6 (ESI†). In each experiment, copper transfer resulted in a rapid decrease in absorbance. All the kinetic data could be fitted by a single exponential function, giving an average rate constant of 268 ± 13 s−1, which was essentially independent of the concentration of CopZ or CopAab, indicative of a true first order process.
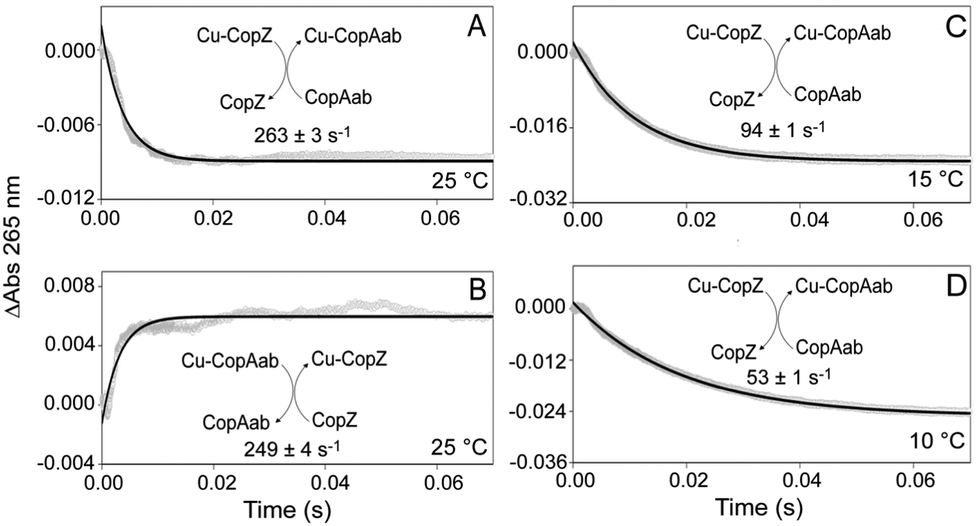 | ||
| Fig. 2 Kinetics of Cu(I) transfer between CopZ and CopAab. Changes in absorbance intensity at 265 nm as a function of time following the mixing of (A) 0.5 Cu/His6CopZ (60 μM) with apo-CopAab (40 μM) and (B) 0.5 Cu/CopAab (60 μM) with apo-CopZ (40 μM) at 25 °C. (C and D) As in (A) but at 15 °C and 10 °C, respectively. The data were fitted with a single exponential function from which an observed rate constant was derived. Proteins were in 100 mM MOPS, 100 mM NaCl, pH 7.5. | ||
Closer inspection of the amplitude of the A265nm changes revealed that they were significantly smaller than those predicted from Fig. S4 (ESI†) on the basis of the absorbance properties of the two proteins for the transfer of half the Cu(I) (as predicted from the thermodynamic transfer experiments in Fig. 1). This suggested that part of the transfer reaction may have occurred during the dead time of the measurement. To investigate this, equivalent experiments were conducted at 15 °C and 10 °C, see Fig. 2C and D. As expected, the rate of the transfer reaction was slower and the amplitude greater, in line with that expected, consistent with all of the transfer reaction being captured at these temperatures. A single exponential function was again used to fit the data, giving rate constants of k = 94 ± 1 s−1 at 15 °C and 53 ± 1 s−1 at 10 °C. Experiments with variable concentrations of Cu(CopZ)2 and apo-CopAab (Fig. S7 and S8, ESI†) revealed that, as at 25 °C, these rate constants are independent of concentration.
To confirm the reversibility of Cu(I)-transfer, Cu(I)–CopAab was mixed with apo-CopZ at 25 °C, see Fig. 2B. This resulted in an increase in absorbance of approximately the same amplitude as that for the decrease in absorbance observed for transfer from CopZ to CopAab. Experiments were conducted with variable protein concentrations (Fig. S9, ESI†) and data fitted with a single exponential, giving a rate constant, k = 260 ± 35 s−1, similar to that for the CopZ to CopAab transfer direction that was also independent of concentration, consistent with a first order reaction. The similar magnitude of changes in A265nm for both directions of transfer is consistent with the approximately equal distribution of Cu(I) between CopZ and CopAab (see Fig. 1) and further demonstrates that Cu(I) transfer is reversible between the proteins.
The data presented here provide, for the first time, a quantitative measure of the rate of transfer of a single Cu(I) ion between a copper chaperone and target protein. It has been proposed that the kinetic barrier to Cu(I) dissociation from the chaperone is reduced through the formation of a (transient) complex between the chaperone and its cognate target protein, in which coordination of Cu(I) by both proteins facilitates dissociation from the chaperone and transfer to the target protein. The formation of such complexes has been reported for several chaperone-transporter soluble domains,22,26,27 including a solution structure of the yeast Atx1/Ccc2a complex.6
To directly detect interaction between CopZ and CopAab, 15N-labeled apo-CopAab was titrated with unlabelled CopZ containing 1 Cu(I) per protein and spectral changes were measured by 1H–15N HSQC experiments. Several 1H and 15N resonances of apo-CopAab showed changes in chemical shift values and/or an increasing broadening (eventually leading to their disappearance) upon addition of increasing amounts of Cu(I)–CopZ (Fig. 3A and Table S1, ESI†). These changes, which are different from those observed on titrating 15N apo-CopAab directly with Cu(I) in the presence of DTT,17 are due to residues close to the Cu(I) ligands of each CopAab domain (CopAa, Cys17, Cys20; CopAb, Cys85 and Cys88), indicating that CopZ simultaneously interacts with both copper-binding sites of CopAab. In particular, the major NMR changes were located in two regions of the protein, which include the two metal binding loops (loop 1 and loop 6), all helices (α1, α2, α3 and α4) and the last loops (loop 5 and loop 10). The exchange rate between free and “interacting” species was larger than, or similar to, the difference in chemical shift, indicating that the proteins in the complex are in fast/intermediate exchange with the free proteins in solution.
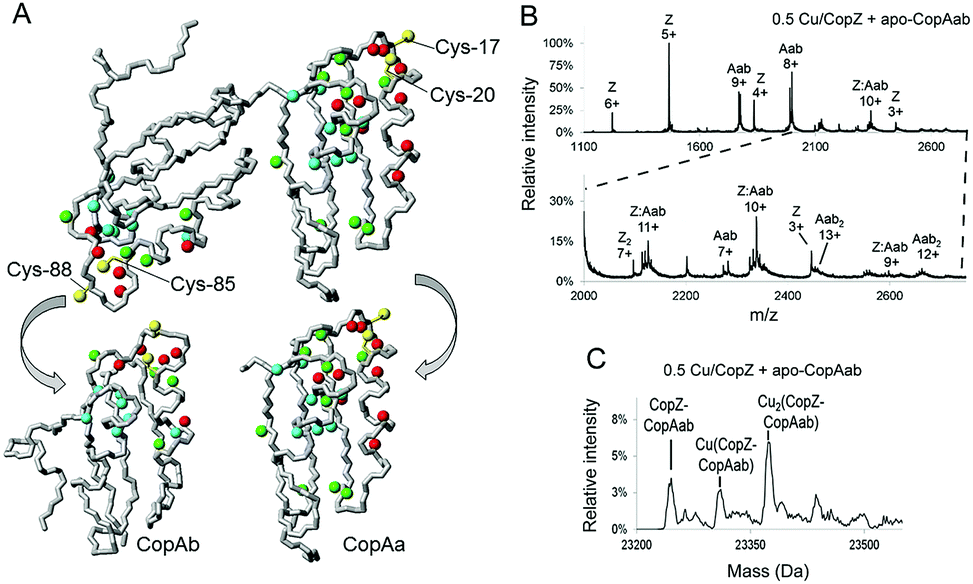 | ||
Fig. 3 The interaction of CopAab with CopZ. (A) 1H, 15N chemical shift changes observed upon interaction of 15N apo-CopAab with unlabelled Cu(I)–CopZ at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio mapped on the apo-CopAab solution structure. NHs cross-peaks broadened beyond detection are shown in red, while those showing measurable changes are shown in green (large NHs chemical shift variations (Δavg(HN) > 0.05 ppm)) or in cyan (small NHs chemical shift variations (0.02 < Δavg(HN) < 0.05 ppm)), where (Δavg(HN) = {[(ΔH)2 + (ΔN/5)2]/2}1/2). Cys residues of both metal binding sites are also shown in yellow. A rotated view of both CopAa and CopAb domains is shown at the bottom to allow for viewing of the interaction surfaces, which are facing the reader. (B) ESI-MS mass spectrum following mixing of 0.5 Cu/CopZ with apo-CopAab. An expanded view of the m/z = 2000–2750 region is shown. Each protein envelope is labelled with the corresponding charge state of each peak, as follows: Aab, CopAab monomer; Aab2, CopAab dimer; Z, CopZ monomer; Z2, CopZ dimer; Z:Aab, CopZ–CopAab complex. (C) Deconvoluted mass spectrum of the CopZ–CopAab complex region for the m/z data shown in (B). Proteins were in 20 mM ammonium acetate, pH 7.4. :2 ratio mapped on the apo-CopAab solution structure. NHs cross-peaks broadened beyond detection are shown in red, while those showing measurable changes are shown in green (large NHs chemical shift variations (Δavg(HN) > 0.05 ppm)) or in cyan (small NHs chemical shift variations (0.02 < Δavg(HN) < 0.05 ppm)), where (Δavg(HN) = {[(ΔH)2 + (ΔN/5)2]/2}1/2). Cys residues of both metal binding sites are also shown in yellow. A rotated view of both CopAa and CopAb domains is shown at the bottom to allow for viewing of the interaction surfaces, which are facing the reader. (B) ESI-MS mass spectrum following mixing of 0.5 Cu/CopZ with apo-CopAab. An expanded view of the m/z = 2000–2750 region is shown. Each protein envelope is labelled with the corresponding charge state of each peak, as follows: Aab, CopAab monomer; Aab2, CopAab dimer; Z, CopZ monomer; Z2, CopZ dimer; Z:Aab, CopZ–CopAab complex. (C) Deconvoluted mass spectrum of the CopZ–CopAab complex region for the m/z data shown in (B). Proteins were in 20 mM ammonium acetate, pH 7.4. | ||
The regions of CopA domain b that were affected were the same as those found in the previously studied interaction of the single domain apo-CopAb with Cu(I)–CopZ, which revealed a 1:1 complex with an affinity of ∼104 M−1.22 Thus, the presence of the first domain does not alter the interaction of the second with CopZ.22 With the exception of Ala9, all residues that are in contact at the interface region between the two domains did not show chemical shift changes, indicating that essentially no structural rearrangements involving relative domain reorientation occur upon interaction of the transporter N-terminal domains with the chaperone. The observed NMR changes described above occurred simultaneously for both domains along all the points of the titration, consistent with the formation of a 2:1 complex, with no preferential interaction of CopZ with respect to the two domains of CopAab.
The interaction between CopZ and CopAab was also investigated by electrospray mass spectrometry (ESI-MS) under non-denaturing conditions (Fig. S10, ESI†). Mass spectra recorded following mixing of 0.5 Cu/CopZ with apo-CopAab and 0.5 Cu/CopAab with apo-CopZ are shown in Fig. 3B and C. Very similar data were obtained when Cu(I) was initially bound to CopAab (Fig. S11 and S12, ESI†), confirming the reversibility of Cu(I) transfer between the two proteins. The spectra contain multiple peak envelopes which arise from the mixture of species in solution: CopZ, CopAab and a complex formed between CopZ and CopAab. The latter is revealed by 9+, 10+ and 11+ charge state envelopes (Fig. 3B and Fig. S11A, ESI†). The deconvoluted mass spectra contain a peak at 23245 Da, consistent with the formation of an adduct of apo-CopZ and apo-CopAab containing two disulfide bonds (predicted 23245 Da). More importantly, two further peaks were observed at ∼+63 and ∼+127 Da, indicating the presence of Cu(CopZ:CopAab) and Cu2(CopZ:CopAab) complexes (Fig. 3C and Fig. S11B, ESI†). The latter species could represent CopAab, which already has one Cu(I) bound at one domain, interacting with a second CopZ at the other domain. The affinity chromatography data, showing that the two proteins can be readily separated from one another, are consistent with weak/transient complex formation.
The bacterial cytoplasm contains millimolar concentrations of low molecular weight thiols; in the case of B. subtilis, this is bacillithiol (BSH).28 We showed recently that BSH inhibits the dimerization of CopZ,21 most likely by out-competing the second CopZ molecule for Cu(I) coordination. However, there is no evidence from structural studies of chaperone-transporter interactions that low molecular weight thiols interfere with, or participate in, complex formation.6,22 We were unable to test the effects of BSH on the transfer kinetics here because of absorbance interference of the thiol that would mask the transfer reaction. However, BSH would not be expected to influence the kinetics of transfer because the rate-limiting step involves rearrangement or resolution of the CopZ–CopAab complex.
Previous kinetic studies of the transfer of Cu(I) from CopZ to the Cu(I)-chelator BCS showed that this is also a first order process, and that the reverse transfer process, from BCS to CopZ, occurs with identical kinetic characteristics, k = ∼0.02 s−1 at 25 °C.20 Thus, Cu(I) transfer between CopZ and CopAab occurs approx. four orders of magnitude more rapidly than between CopZ and BCS, consistent with the physiological importance of the CopZ–CopAab transfer reaction. These exchange processes have in common that the rate-limiting step is the same for both directions of transfer and is a first order process. Thus, it cannot correspond to the initial binding of CopAab to CopZ, which is a second order process. Quantum mechanics/molecular mechanics (QM/MM) studies of Cu(I) transfer from Atox1 to the fourth MBD of Wilson disease protein concluded that significant activation barriers were associated with the transition from coordination by two chaperone/one transporter Cys residues to one chaperone/two transporter Cys residues, and also with the decoordination of the chaperone.29 This is consistent with our data and we propose that the rate limiting step of transfer corresponds to either reorganisation of Cys thiolate coordination within, or the dissociation of, a transient CopZ–Cu–CopAab complex. We note that previous NMR studies of the interaction between CopZ and CopAa indicated a rate constant for the dissociation of the CopZ–CopAa complex of ∼600 s−1,22 again consistent with our observations. The rate constant determined here effectively represents that for the CopAab-mediated dissociation of Cu(I) from CopZ. This is ten orders of magnitude higher than that predicted (see above) and at least seven orders higher than that estimated here for Cu(I) dissociation in the absence of CopAab.
In summary, the data presented here demonstrates the reversible transfer of Cu(I) between CopZ and CopAab, mediated by complex formation between the proteins. The rate at which transfer occurs indicates a remarkable enhancement of the rate constant for dissociation of Cu(I) from CopZ of many orders of magnitude compared to when CopAab is absent. This demonstrates that complex formation between chaperone and target protein functions to dramatically lower the kinetic barrier to Cu(I) dissociation from the chaperone.
This work was supported by UEA through the award of a studentship to K. K., financial support to L. Z., and through the purchase of the mass spectrometer in our lab, by the UK's BBSRC (through the award of studentships to C. S. and M. A. K.), by the European Commission (Contract no. 026145), and by Marie Curie Host Fellowships (no. MEST-CT-2004-504391).
Notes and references
- A. C. Rosenzweig, Acc. Chem. Res., 2001, 34, 119–128 CrossRef CAS PubMed.
- C. Singleton and N. E. Le Brun, Biometals, 2007, 20, 275–289 CrossRef CAS PubMed.
- R. A. Pufahl, C. P. Singer, K. L. Peariso, S.-J. Lin, P. J. Schmidt, C. J. Fahrni, V. Cizewski Culotta, J. E. Penner-Hahn and T. V. O'Halloran, Science, 1997, 278, 853–856 CrossRef CAS PubMed.
- F. Arnesano, L. Banci, I. Bertini, D. L. Huffman and T. V. O'Halloran, Biochemistry, 2001, 40, 1528–1539 CrossRef CAS PubMed.
- F. Arnesano, L. Banci, I. Bertini, F. Cantini, S. Ciofi-Baffoni, D. L. Huffman and T. V. O'Halloran, J. Biol. Chem., 2001, 276, 41365–41376 CrossRef CAS PubMed.
- L. Banci, I. Bertini, F. Cantini, I. C. Felli, L. Gonnelli, N. Hadjiliadis, R. Pierattelli, A. Rosato and P. Voulgaris, Nat. Chem. Biol., 2006, 2, 367–368 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, D. L. Huffman and T. V. O'Halloran, J. Biol. Chem., 2001, 276, 8415–8426 CrossRef CAS PubMed.
- M. E. Portnoy, A. C. Rosenzweig, T. Rae, D. L. Huffman, T. V. O'Halloran and V. C. Culotta, J. Biol. Chem., 1999, 274, 15041–15045 CrossRef CAS PubMed.
- L. Banci, I. Bertini, V. Calderone, N. Della-Malva, I. C. Felli, S. Neri, A. Pavelkova and A. Rosato, Biochem. J., 2009, 422, 37–42 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Nature, 2010, 465, 645–648 CrossRef CAS PubMed.
- D. L. Huffman and T. V. O'Halloran, J. Biol. Chem., 2000, 275, 18611–18614 CrossRef CAS PubMed.
- D. S. Radford, M. A. Kihlken, G. P. M. Borrelly, C. R. Harwood, N. E. Le Brun and J. S. Cavet, FEMS Microbiol. Lett., 2003, 220, 105–112 CrossRef CAS PubMed.
- L. Banci, I. Bertini, R. Del Conte, J. Markey and F. J. Ruiz-Duenas, Biochemistry, 2001, 40, 15660–15668 CrossRef CAS PubMed.
- M. A. Kihlken, A. P. Leech and N. E. Le Brun, Biochem. J., 2002, 368, 729–739 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, L. Gonnelli and X.-C. Su, J. Biol. Chem., 2003, 278, 50506–50513 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, L. Gonnelli and X.-C. Su, J. Mol. Biol., 2003, 331, 473–484 CrossRef CAS PubMed.
- C. Singleton, L. Banci, S. Ciofi-Baffoni, L. Tenori, M. A. Kihlken, R. Boetzel and N. E. Le Brun, Biochem. J., 2008, 411, 571–579 CrossRef CAS PubMed.
- C. Singleton and N. E. Le Brun, Dalton Trans., 2009, 688–696 RSC.
- S. Hearnshaw, C. West, C. Singleton, L. Zhou, M. A. Kihlken, R. W. Strange, N. E. Le Brun and A. M. Hemmings, Biochemistry, 2009, 48, 9324–9326 CrossRef CAS PubMed.
- L. Zhou, C. Singleton and N. E. Le Brun, Biochem. J., 2008, 413, 459–465 CrossRef CAS PubMed.
- K. L. Kay, C. J. Hamilton and N. E. Le Brun, Metallomics, 2016, 8, 709–719 RSC.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, R. Del Conte and L. Gonnelli, Biochemistry, 2003, 42, 1939–1949 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, M. D'Onofrio, L. Gonnelli, F. C. Marhuenda-Egea and F. J. Ruiz-Duenas, J. Mol. Biol., 2002, 317, 415–429 CrossRef CAS PubMed.
- P. Gourdon, X. Y. Liu, T. Skjorringe, J. P. Morth, L. B. Moller, B. P. Pedersen and P. Nissen, Nature, 2011, 475, 59–64 CrossRef CAS PubMed.
- F. J. Enguita, L. O. Martins, A. O. Henriques and M. A. Carrondo, J. Biol. Chem., 2003, 278, 19416–19425 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, C. T. Chasapis, N. Hadjiliadis and A. Rosato, FEBS J., 2005, 272, 865–871 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, N. G. Kandias, N. J. Robinson, G. A. Spyroulias, X.-C. Su, S. Tottey and M. Vanarotti, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8320–8325 CrossRef CAS PubMed.
- G. L. Newton, M. Rawat, J. J. La Clair, V. K. Jothivasan, T. Budiarto, C. J. Hamilton, A. Claiborne, J. D. Helmann and R. C. Fahey, Nat. Chem. Biol., 2009, 5, 625–627 CrossRef CAS PubMed.
- A. Rodriguez-Granillo, A. Crespo, D. A. Estrin and P. Wittung-Stafshede, J. Phys. Chem. B, 2010, 114, 3698–3706 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc08966f |
| This journal is © The Royal Society of Chemistry 2017 |