
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh oxidation state bromocarbyne complexes†
Anthony F.
Hill
 * and
Richard Y.
Kong
* and
Richard Y.
Kong
Research School of Chemistry, Australian National University, Canberra, Australian Capital Territory ACT 2601, Australia. E-mail: a.hill@anu.edu.au
First published on 12th December 2016
Abstract
Bromination of the carbyne complexes [W(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)Br(CO)2(dcpe)] (R = Ph, SiPh3; dcpe = 1,2-bis(dicyclohexylphosphino)ethane) provides high oxidation state derivatives [W(CPh)Br3(dcpe)] and [W(CBr)Br3(dcpe)], the latter via an unprecedented bromodesilylation process.
CR)Br(CO)2(dcpe)] (R = Ph, SiPh3; dcpe = 1,2-bis(dicyclohexylphosphino)ethane) provides high oxidation state derivatives [W(CPh)Br3(dcpe)] and [W(CBr)Br3(dcpe)], the latter via an unprecedented bromodesilylation process.
In 1983 Lalor reported the first halocarbyne complexes [M(
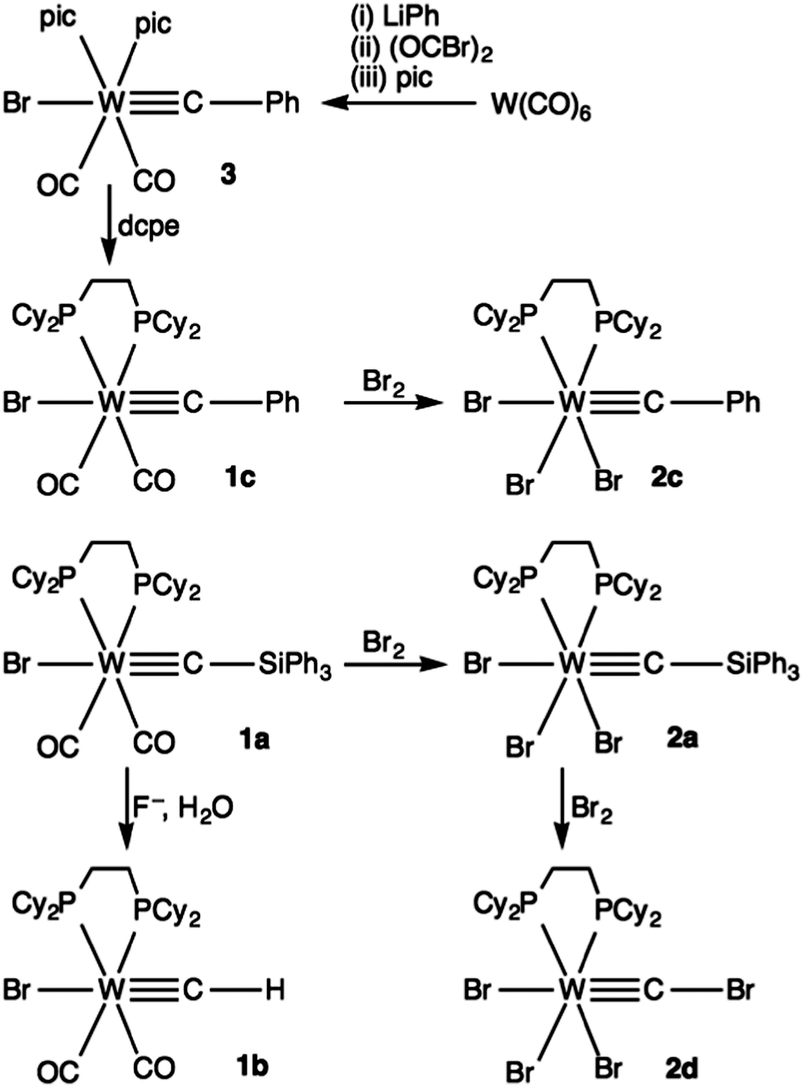
CX)(CO)2(Tp*)] (M = Mo, W; X = Cl, Br; Tp* = hydrotris(dimethylpyrazolyl)borate).1 In the interim these have been proven to be remarkably versatile building blocks for the construction of elaborate and otherwise inaccessible carbyne derivatives via either nucleophilic substitution (spontaneous or Pd0-mediated)2 or alternatively, lithium–halogen exchange to afford nucleophilic lithiocarbynes [M(CLi)(CO)2(Tp*)] (Scheme 1).3 Such protocols are however denied wider generality in that some 33 years later Lalor's complexes still remain the only known halocarbynes with the exception of Hughes' fluorocarbynes [M(CF)(CO)2(L)] (M = Cr, Mo, W; L = η-C5R5, R = H, Me)4 for which no subsequent reactivity has been reported. Extension of Lalor's synthetic approach involving the reaction of the carbonylates [M(CO)3(Tp*)]− with diazonium salts in haloform solvents, to any other ligand sets or metals has not met with success despite considerable effort.1b We have therefore considered alternative approaches to the synthesis of MC1(±) synthons so as to broaden the applicability of the methodologies outlined in Scheme 1. In this pursuit, we have recently shown that fluoride-mediated protodesilyation of the silylcarbyne complex [W(CSiPh3)Br(CO)2(dcpe)] [(1a,‡ dcpe) = 1,2-bis(dicyclohexylphosphino)ethane] affords a rare example of a remarkably stable ‘parent’ methylidyne complex [W(CH)Br(CO)2(dcpe)] (1b, Scheme 2).5
 | ||
| Scheme 1 Umpolung of bromocarbyne reactivity (Nu = nucleophile, E = electrophile, M = Mo, W). | ||
 | ||
| Scheme 2 | ||
Roper first showed that it was possible to oxidise carbyne complexes without compromising the metal–carbon triple bond5 and this was further developed by Mayr who demonstrated the bromination of Fischer's carbyne [W(CPh)Br(CO)4] in dimethoxyethane (dme) to afford the Schrock-type carbyne [W(CPh)Br3(dme)].6 We have therefore explored the possibility of preparing a high-oxidation state§ silylcarbyne fac-[W(CSiPh3)Br3(dcpe)] (2a) by bromination of 1a.
Prior to the pursuit of 2a, we first validated the approach by brominating the new but more conventional benzylidyne complex [W(CPh)Br(CO)2(dcpe)] (1c, Scheme 2), obtained by a classical Fischer-Mayr oxide abstraction route.7 Thus treating W(CO)6 sequentially with phenyl lithium, oxalyl bromide, 4-picoline and dcpe provides 1c (see Fig. 1 and ESI† for characterisational data). Treating 1c with bromine at low temperature results in the formation of the high-valent tungsten benzylidyne complex fac-[W(CPh)Br3(dcpe)] (2c, 98%, Fig. 2). The 13C resonance for the benzylidyne appears at δC = 309.7 (2JCP = 13.4 Hz), moved some 46 ppm to lower frequency from that for 1c (δC = 263.7, 2JCP = 9.3 Hz). The 31P resonance at 53.1 ppm shows satellite resonances arising from coupling to 183W (1JWP = 212 Hz) which is comparable in magnitude to that observed for 1c (225 Hz).
 | ||
| Fig. 1 Molecular structure of 1c in a crystal (60% displacement ellipsoids, H-atoms, cyclohexyl groups simplified). Selected bond lengths (Å) and angles (°): W1–P1 2.5318(6), W1–P2 2.5407(6), W1–Br 2.6929(3), W1–C1 1.821(3), P1–W1–P2 80.38(2), C1–W1–P1 93.97(8), C1–W1–P2 97.55(8), C1–W1–Br1 171.46(8), C2–C1–W1 171.6(2). | ||
 | ||
| Fig. 2 Molecular structure of 2c in a crystal (60% displacement ellipsoids, H-atoms and solvent omitted, cyclohexyl groups simplified, one of two crystallographically independent molecules shown). Selected bond lengths (Å) and angles (°): W1–Br3 2.7549(5), W1–Br4 2.4914(5), W1–Br6 2.4782(5), W1–P9 2.5786(11), W1–C23 1.789(5), C34–C23 W1 174.2(4). | ||
With the oxidative dibromination of 1c to provide 2c having proceeded without issue, the corresponding reaction with 1a was explored. Addition of bromine to a solution of 1a at −78 °C followed by slow warming to room temperature resulted in the formation of a khaki complex, the yield of which is optimised by employing two equivalents of Br2. The product was formulated on the basis of spectroscopic (see ESI†) and structural data (Fig. 3) as the bromocarbyne complex [W(CBr)Br3(dcpe)] (2d) resulting from both oxidation of the metal centre and electrophilic bromodesilylation of the silylcarbyne ligand (Scheme 2).
 | ||
| Fig. 3 Molecular structure of 2d in a crystal of 2d·CH2Cl2 (60% displacement ellipsoids, H-atoms and solvent omitted, cyclohexyl groups simplified, one of two crystallographically independent molecules shown). Selected bond lengths (Å) and angles (°): W2–Br3 2.6918(6), W2–Br5 2.5016(7), W2–Br6 2.4800(7), W2–P3 2.5862(16), W2–P4 2.5734(16), W2–C1 1.793(6), Br1–C1 1.822(6), P4–W2–P3 76.35(5), W2–C1–Br1 171.5(4). | ||
The key spectroscopic datum of interest is the triplet 13C resonance observed at δC = 229.0 which shows coupling (2JCP = 16.4 Hz) approximately twice the magnitude of those found for lower valent dcpe tungsten carbyne complexes. Within the low-valent regime, complexes of the form [M(CR)(CO)2(L)] (M = Cr, Mo, W; L = HB(pzR2-3,5)3 R = H, Me)8 provide the most extensive range of comparative spectroscopic data. Of these, the bromocarbyne complexes have characteristically high frequency resonances (e.g., [M(CBr)(CO)2(Tp*)]: δC = 198.0 M = W; 202.5 M = Mo), exceeded only by the iodocarbyne [W(CI)(CO)2(Tp*)] (δC = 183.2).9 Thus the ca. 81 ppm shift to high frequency of the carbyne resonance for 2d relative to 2c is to be expected. The infrared spectrum included a comparatively strong absorption at 1260 cm−1 which on the basis of simulation (DFT: M06-LACVP) for the simpler model [W(CBr)Br3(dmpe)] (νWC = 1203 cm−1) we assign to the primarily νWC mode. No such ‘pure’ mode could be identified for the corresponding benzylidyne complex due to extensive coupling with phenyl ring modes. Infrared data for carbyne ligands are somewhat sparse in the literature and this may be traced, with good reason, to a number of factors. Firstly, they appear in the fingerprint region amongst co-ligand absorptions. The polarity of MC bonds (and thus changes in dipole moments associated with IR intensities) can vary dramatically, hence the Fischer–Schrock dichotomy. Finally, and most importantly, in contrast to metal oxo and nitride ligands, for which IR data are invaluable and unambiguous, the MC oscillator is invariably coupled to modes due to the substituent, a phenomenon that has been discussed in detail by Dao et al.10 These factors taken together mean that the value of νMC covers a wide range of both frequency and intensity such that reliable assignments are best made with recourse to simulation.
The molecular geometry of 2d (Fig. 3) involves pseudo-octahedral tungsten with a typically short W2–C1 bond length of 1.793(6) Å that is not however significantly different (1 e.s.d.) to that found in 2c. Carbyne ligands typically exert a significant trans influence and this is reflected in the W2–Br3 bond length (2.6918(6) Å) being significantly longer than those trans to phosphorus (2.5016(7), 2.4800(7) Å), but less elongated relative to the corresponding bromide in 2c (126 e.s.d.), suggesting that the bromocarbyne ligand, for which no previous structural data are available, has a pronounced trans influence, though less than that for more conventional alkylidynes. The C1–Br1 bond length of 1.822(6) Å is somewhat longer than any previously reported C(sp)–Br bonds which span the range 1.77–1.80 Å. The acute dcpe bite angle of 76.35(5)° allows the cis-bromo ligands to adopt a significantly obtuse angle (109.35(2)°).
The availability of both 2c and 2d provides an opportunity to compare these disparate carbyne substituents. Computational interrogation of the models [W(CR)Br3(dmpe)] (R = Ph, Br, DFT: MO6 LACVP) indicates that replacing the phenyl substituent in 2c with a bromide in 2d results in an increase in the negative (natural atomic) charge on the carbyne carbon (−0.29 to −0.51). The positive charge on tungsten is essentially unchanged for the benzylidyne (+0.75) and bromocarbyne (+0.78) models.
The stepwise mechanism by which 1a is converted to 2d would appear to involve initial oxidative decarbonylation of the tungsten followed by slower bromo-desilylation of the silylcarbyne. We assert this sequence based on the following observations. (i) CO evolution is immediately observed at low temperature; (ii) bromination of [W(CPh)Br(CO)2(dcpe)] (1c) cleanly affords the benzylidyne complex [W(CPh)Br3(dcpe)] (2c). As to the intimate mechanism for C–Si bond cleavage, in an isolobal context Isobe provided the protocol of choice for the electrophilic bromination of alkynylsilanes by N-bromosuccinimide to afford alkynyl bromides,11 however a silver co-catalyst (AgNO3, AgF) is required, implicating silver alkynyl intermediates. In the conversion of 1a to 2d however direct attack by Br2 presumably affords transient [W(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CBrSiPh3)Br3(dcpe)]Br with subsequent attack at silicon by bromide. Electrophilic C-halogenation of carbyne ligands has been described previously, but only in one instance (chlorination of [Os(CR)Cl(CO)(PPh3)2] to afford [Os(CClR)Cl2(CO)(PPh3)2], R = C6H4Me-4)12 and whilst bromocarbene complexes13,14 are rare and highly reactive due to the exceptional halide nucleofugacity, the complex [Re2(CBrSiPh3)(CO)9]15 provides limited precedent.
CBrSiPh3)Br3(dcpe)]Br with subsequent attack at silicon by bromide. Electrophilic C-halogenation of carbyne ligands has been described previously, but only in one instance (chlorination of [Os(CR)Cl(CO)(PPh3)2] to afford [Os(CClR)Cl2(CO)(PPh3)2], R = C6H4Me-4)12 and whilst bromocarbene complexes13,14 are rare and highly reactive due to the exceptional halide nucleofugacity, the complex [Re2(CBrSiPh3)(CO)9]15 provides limited precedent.
In conclusion, a considerable number of silylcarbyne complexes are available via classical Fischer alkoxide/oxide abstraction,5,15,16 Schrock α-hydrogen elimination/abstraction17 or alkynylsilane metathesis18 protocols. The demonstration that halodesilylation may be achieved without rupture of the metal-carbon multiple bond augurs well for the elaboration of further synthetically valuable halocarbyne complexes to open the 33 year hiatus in their wider exploration.
Notes and references
- (a) T. Desmond, F. J. Lalor, G. Ferguson and M. Parvez, Chem. Commun., 1983, 457–458 RSC; (b) T. J. Desmond, F. J. Lalor, G. Ferguson, M. Parvez and T. Wieckowski, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 59–61 CrossRef; (c) F. J. Lalor, T. J. Desmond, G. M. Cotter, C. A. Shanahan, G. Ferguson, M. Parvez and B. Ruhl, J. Chem. Soc., Dalton Trans., 1995, 1709–1726 RSC.
- (a) T. Desmond, F. J. Lalor, G. Ferguson and M. Parvez, J. Chem. Soc., Chem. Commun., 1984, 75–77 RSC; (b) F. J. Lalor and S. A. O'Neill, J. Organomet. Chem., 2003, 684, 249–265 CrossRef CAS; (c) S. Chaona, F. J. Lalor, G. Ferguson and M. M. Hunt, J. Chem. Soc., Chem. Commun., 1988, 1606–1608 RSC; (d) L. Weber, G. Dembeck, R. Boese and D. Blaeser, Organometallics, 1999, 18, 4603–4607 CrossRef CAS; (e) L. Weber, G. Dembeck, R. Boese and D. Blaser, Chem. Ber., 1997, 130, 1305–1308 CrossRef CAS; (f) B. E. Woodworth and J. L. Templeton, J. Am. Chem. Soc., 1996, 118, 7418–7419 CrossRef CAS; (g) G. M. Jamison, P. S. White and J. L. Templeton, Organometallics, 1991, 10, 1954–1959 CrossRef CAS; (h) M. Etienne, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1991, 113, 2324–2325 CrossRef CAS; (i) M. I. Bruce, M. L. Cole, M. Gaudio, B. W. Skelton and A. H. White, J. Organomet. Chem., 2006, 691, 4601–4614 CrossRef CAS; (j) D. J. Armitt, M. I. Bruce, M. Gaudio, N. N. Zaitseva, B. W. Skelton, A. H. White, B. Le Guennic, J.-F. Halet, M. A. Fox, R. L. Roberts, F. Hartl and P. J. Low, Dalton Trans., 2008, 6763–6775 RSC; (k) R. L. Cordiner, P. A. Gugger, A. F. Hill and A. C. Willis, Organometallics, 2009, 28, 6632–6635 CrossRef CAS; (l) R. L. Cordiner, A. F. Hill and J. Wagler, Organometallics, 2008, 27, 4532–4540 CrossRef CAS; (m) L. M. Caldwell, A. F. Hill, A. D. Rae and A. C. Willis, Organometallics, 2008, 27, 341–345 CrossRef CAS; (n) L. M. Caldwell, A. F. Hill, R. Stranger, R. N. L. Terrett, K. M. von Nessi, J. S. Ward and A. C. Willis, Organometallics, 2015, 34, 328–334 CrossRef CAS; (o) A. L. Colebatch and A. F. Hill, J. Am. Chem. Soc., 2014, 136, 17442–17445 CrossRef CAS PubMed.
- (a) R. L. Cordiner, A. F. Hill and J. Wagler, Organometallics, 2008, 27, 5177–5179 CrossRef CAS; (b) A. L. Colebatch and A. F. Hill, Organometallics, 2016, 35, 2249–2255 CrossRef CAS; (c) A. L. Colebatch, A. F. Hill and M. Sharma, Organometallics, 2015, 34, 2165–2182 CrossRef CAS; (d) E. S. Borren, A. F. Hill, R. Shang, M. Sharma and A. C. Willis, J. Am. Chem. Soc., 2013, 135, 4942–4945 CrossRef CAS PubMed; (e) A. F. Hill and R. Shang, Organometallics, 2012, 31, 4635–4638 CrossRef CAS; (f) A. F. Hill, M. Sharma and A. C. Willis, Organometallics, 2012, 31, 2538–2542 CrossRef CAS; (g) A. F. Hill, R. Shang and A. C. Willis, Organometallics, 2011, 30, 3237–3241 CrossRef CAS; (h) A. L. Colebatch, A. F. Hill, R. Shang and A. C. Willis, Organometallics, 2010, 29, 6482–6487 CrossRef CAS.
- (a) H. Huang, R. P. Hughes, C. R. Landis and A. L. Rheingold, J. Am. Chem. Soc., 2006, 128, 7454–7455 CrossRef CAS PubMed; (b) H. Huang, R. P. Hughes and A. L. Rheingold, Dalton Trans., 2011, 40, 47–55 RSC.
- A. F. Hill, J. S. Ward and Y. Xiong, Organometallics, 2015, 34, 5057–5064 CrossRef CAS.
- (a) G. R. Clark, N. R. Edmonds, R. A. Pauptit, W. R. Roper, J. M. Waters and A. H. Wright, J. Organomet. Chem., 1983, 244, C57–C60 CrossRef CAS; (b) W. R. Roper, J. Organomet. Chem., 1986, 300, 167–190 CrossRef CAS.
- A. Mayr and G. A. McDermott, J. Am. Chem. Soc., 1986, 108, 548–549 CrossRef CAS PubMed.
- L. M. Caldwell, Adv. Organomet. Chem., 2008, 56, 1–94 CrossRef CAS.
- A. E. Enriquez, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 2001, 123, 4992–5002 CrossRef CAS PubMed.
- N. Q. Dao, E. O. Fischer and C. Kappenstein, Nouv. J. Chim., 1980, 4, 85–94 Search PubMed.
- T. Nishikawa, S. Shibuya, S. Hosokawa and M. Isobe, Synlett, 1994, 485–486 CrossRef CAS.
- G. R. Clark, K. Marsden, W. R. Roper and L. J. Wright, J. Am. Chem. Soc., 1980, 102, 6570–6571 CrossRef CAS.
- For a review of halomethyl and halomethylidene complexes see P. J. Brothers and W. R. Roper, Chem. Rev., 1998, 88, 1293–1326 CrossRef.
- (a) T. G. Richmond, A. M. Crespi and D. F. Shriver, Organometallics, 1984, 3, 314–319 CrossRef CAS; (b) A. M. Crespi and D. F. Shriver, Organometallics, 1985, 4, 1830–1835 CrossRef CAS; (c) Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Chem. – Eur. J., 2010, 16, 2240–2249 CrossRef CAS PubMed; (d) D. Mansuy, Pure Appl. Chem., 1982, 54, 638–639 Search PubMed; (e) E. O. Fischer, W. Kleine and F. R. Kreißl, J. Organomet. Chem., 1976, 107, C23–C25 CrossRef CAS.
- E. O. Fischer, P. Rustemeyer and K. Ackermann, Chem. Ber., 1982, 115, 3851–3859 CrossRef CAS.
- (a) E. O. Fischer, H. Hollfelder, P. Friedrich, F. R. Kreißl and G. Huttner, Angew. Chem., Int. Ed. Engl., 1977, 16, 401–402 CrossRef; (b) J. C. Jeffery, M. A. Ruiz and F. G. A. Stone, J. Organomet. Chem., 1988, 355, 231–242 CrossRef CAS; (c) G. M. Jamison, A. E. Bruce, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1991, 113, 5057–5059 CrossRef CAS; (d) R. L. Cordiner, A. F. Hill, R. Shang and A. C. Willis, Organometallics, 2011, 30, 139–144 CrossRef CAS.
- (a) P. D. Savage, G. Wilkinson, M. Motevalli and M. B. Hursthouse, Polyhedron, 1987, 6, 1599–1601 CrossRef CAS; (b) L. A. Morton, M. Miao, T. M. Callaway, T. Chen, S.-J. Chen, A. A. Tuinman, X. Yu, Z. Lu and Z.-L. Xue, Chem. Commun., 2013, 49, 9555–9557 RSC; (c) L. Giannini, E. Solari, S. Dovesi, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1999, 121, 2784–2796 CrossRef CAS; (d) G. Balazs, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2005, 44, 4920–4924 CrossRef CAS PubMed; (e) A. S. Hock and R. R. Schrock, Chem. – Asian J., 2007, 2, 867–874 CrossRef CAS PubMed; (f) S. W. Seidel, R. R. Schrock and W. M. Davis, Organometallics, 1998, 17, 1058–1068 CrossRef CAS.
- (a) M. L. Listemann and R. R. Schrock, Organometallics, 1985, 4, 74–83 CrossRef CAS; (b) A. V. Safronova, L. N. Bochkarev, N. E. Stolyarova, I. K. Grigor'eva, I. P. Malysheva, G. V. Basova, G. K. Fukin, Y. A. Kursky, S. Y. Khorshev and G. A. Abakumov, Russ. Chem. Bull., 2003, 2140–2145 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic and crystallographic data, the latter in .cif format. CCDC 1515752–1515754. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08946a |
| ‡ Hereafter in compound numbers a = SiPh3, b = H, c = Ph, d = Br. |
| § Throughout, we employ the [CR]3− formalism for electron counting and the assignment of oxidations states and associated d-configurations. This is an arbitrary distinction in such highly covalent compounds and the alternative [CR]+ formalism also has advocates. |
| This journal is © The Royal Society of Chemistry 2017 |