
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional small-molecules & polymers containing P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C and AsC bonds as hybrid π-conjugated materials†
C and AsC bonds as hybrid π-conjugated materials†
Daniel
Morales Salazar
 a,
Edgar
Mijangos
a,
Sonja
Pullen
a,
Ming
Gao
b and
Andreas
Orthaber
*a
a,
Edgar
Mijangos
a,
Sonja
Pullen
a,
Ming
Gao
b and
Andreas
Orthaber
*a
aDepartment of Chemistry, Ångström Laboratories, Molecular Inorganic Chemistry, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: andreas.orthaber@kemi.uu.se
bDepartment of Chemistry, Ångström Laboratories, Polymer Chemistry, Uppsala University, Sweden
First published on 22nd December 2016
Abstract
Stable phospha- and arsaalkenes were used to synthesize polymers containing unsaturated P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and AsC moieties. The composition, chemical environment, structure, optical, and electronic properties of the monomers and polymers were elucidated. The incorporation of the heteroatom–carbon double bonded units efficiently perturbs the optoelectronics and solid state features of both monomeric and polymeric scaffolds. Proof-of principle work supports their responsive character through post-functionalisation and electrochromic behaviour. To the best of our knowledge, this is the first example of a polymer containing arsenic–carbon double bonds.
C and AsC moieties. The composition, chemical environment, structure, optical, and electronic properties of the monomers and polymers were elucidated. The incorporation of the heteroatom–carbon double bonded units efficiently perturbs the optoelectronics and solid state features of both monomeric and polymeric scaffolds. Proof-of principle work supports their responsive character through post-functionalisation and electrochromic behaviour. To the best of our knowledge, this is the first example of a polymer containing arsenic–carbon double bonds.
The diverse bonding and electronic characteristics offered by heavier main group elements (e.g. Si, Ge, P, As, S, Se, etc.) provide a powerful strategy to modify the optical, electronic and solid state properties of π-conjugated systems.1 Within Group 15, the presence of a lone pair in heavier pnictogens orthogonal to the π-system offers a unique possibility for further property tailoring;2 examples such as luminescent poly-2,5-dithienylphosphole/oxides (A), electrochromic 2,7-diaza-dibenzophosphole-oxides (B),3 or the recently reported arsole-containing co-polymer as an oxygen insensitive p-type semiconducting material (C),4 illustrate the flexibility and differences of using congener atoms (Fig. 1). Unlike phospholes, the utilization of acyclic π-conjugated materials including heavier main group elements is limited.5–7 Both poly-(p-phenylenephosphine)s (D)5–7 and poly-(vinylenearsine)s (E)8 have been reported as heavier examples of polyanilines, however the pyramidal nature of As and P inhibits effective π-conjugation. Heavier analogues of alkenes with σ3λ2-P-centres, such as poly(p-phenylenephosphaalkene) (F)9 were shown to lose conformational planarity due to sterically demanding protecting groups needed to kinetically stabilize the P
C bond. Due to the high reactivity of compounds containing unsaturated AsC bonds,10 no polymers containing arsaalkenes have been reported to date. The incorporation of arsenic into π-conjugated hybrid systems is thus stimulating; its polarizable nature, lower LUMO levels leading to higher electron affinity,11 lower oxophilicity in comparison to phosphorus species, diffuse frontier orbital nature that could be exploited in a stimuli responsive fashion, as well as innate atomic properties which could be used in a perturbative manner, are a few hypotheses for its usage.
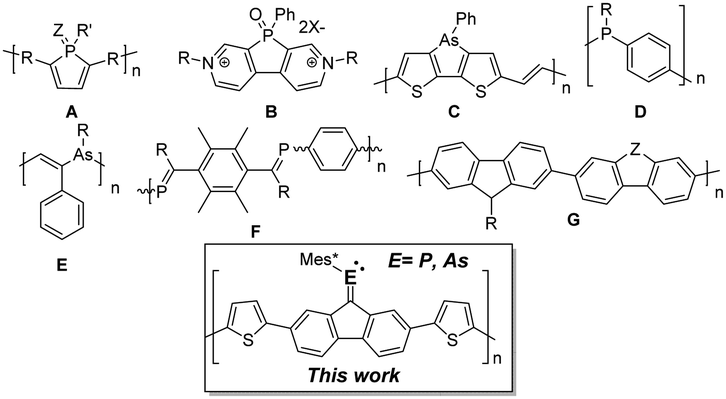 | ||
| Fig. 1 Selected examples of polymer and molecular materials containing pnictogens (A–F) and polyfluorene (G); Mes* = 2,4,6-tri-tert-butylphenyl. | ||
Fluorene based organic materials are heavily investigated because of their versatile optical and electronic properties; to this end, the main-group element approach into fluorene derivatives focuses in creating co-polymers with heterocyclic motifs (e.g. phospholes, thiophenes) or in substituting the fluorenylidene–carbon bridge (G) with a heavy atom (Z = SiR2, PR, S).12 Our goal toward molecular motifs containing co-planar EC units motivated us to synthesize phosphaalkene (poly-2a) and arsaalkene (poly-2b) π-conjugated polymers (Scheme 1). In order to support the functionality of these materials, both polymers were used to show reversible electrochromism; moreover, coordination of 2a and poly-2a to Au(I) ions induced extensive optical and electronic changes of the resulting materials.
 | ||
| Scheme 1 Synthesis of phosphaalkenes (1a, 2a), arsaalkenes (1b, 2b), and polymers poly-2a and poly-2b. (i) (1) n-BuLi, (2) Mes*ECl2, (3) DBU; (ii) Stille coupling; (iii) electropolymerization. | ||
The thienyl-functionalized phosphaalkene 2a and arsaalkene 2b were obtained by a microwave assisted Stille coupling reaction of 1a and 1b with 2-(tributylstannyl)thiophene (Scheme 1). Compounds 2a and 2b were characterized by elemental analysis, NMR (1H, 13C, and 31P), UV-Vis, X-ray diffraction, and high-resolution mass spectroscopy.
Notably, the synthesized compounds are indefinitely air-stable in the solid state and in solution under inert conditions. The 13C-NMR chemical shift corresponding to the pnictogen–carbon double bond (1aδ: 166.7, 2aδ: 167.7, 1bδ: 180.6, 2bδ: 182.0) increases going from phosphorus to arsenic, which indicates higher electron density at the carbon (i.e. ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) E) of 2a compared to 2b. The 31P-NMR spectra of 1a and 1b display low-field signals at 273.7 and 264.9 ppm, which are characteristic of non-polarized PC bonds and phosphaalkenes in delocalized π-conjugated systems. Structure analysis reveals typical PC and AsC bond lengths of 1.693(5) (1a), 1.677(5) (2a) and 1.807(3) Å (1b), respectively (Fig. S14, ESI†). The monomers have a coplanar fluoren-9-ylidene pnictogen backbone (the heteroatoms are only 0.061(1) (1a), 0.063(1) (1b) and −0.007(2) (2a) above/below the least squares plane of the fluorene core). The UV-Vis spectra of 1a and 1b show lowest-energy absorption bands at 371 and 398 nm, respectively (Fig. 2a). In comparison, the λmax of fluorene derivatives such as 2,7-di-bromo-9H-fluorene and 2,7-dibromo-9-methylene-9H-fluorene occurs at 311 and 312 nm. In comparison to all-carbon analogues, the introduction of the pnictinidene unit causes substantial changes in the optical spectrum as well as in the electronic structure of the system, as evidenced by the calculated excited-state transitions (see Fig. S1, ESI†). Dithienyl derivative 2a shows an additional shoulder at 399 nm (Fig. 2a) and a red-shifted broad band centred at 485 nm (λonset 552 nm).13 Similarly, 2b displays an absorption band at 409 nm and a low-energy band centred at around 530 nm (λonset 596 nm); the red-shifted features are due to extended conjugation on the π-backbone through thienyl-functionalization. The frontier molecular orbitals of 2a and 2b show the similarities between P and As (Fig. S2, ESI†), their HOMO corresponds to delocalized π-orbitals across the fluorenylidene-thienyl backbone, whereas their LUMO to fulvene-core π* orbitals, with a dominant contribution of the pnictogen–carbon double bond, which is more stabilized for 2b compared to 2a. The red shifts going from phosphinidene to arsinidene are mainly due to LUMO stabilization (as well as HOMO−1 destabilization). The HOMO−3 displays some lone pair contribution accessible for further functionalization (e.g. metal coordination, vide infra), which could be investigated in sensor devices.14 Electron density difference maps (EDDMs) from TD-DFT calculations provide insights on the difference in electron density for the low-energy transitions (Fig. 2a and Fig. S2, ESI†). The lowest-energy feature corresponds to a HOMO–LUMO transition with charge transfer character from the thienyl fluorene backbone to the hetero-fulvenoid core. The transitions between 350 and 415 nm are mainly based on π–π* transitions involving the fulvenoid pnictogen–carbon antibonding orbitals, which confirms that the phosphaalkene and arsaalkene are intrinsic to the molecular electronic structure of the systems. The electrochemical properties of the monomers were investigated using cyclic voltammetry (Fig. 2b). Both monomers showed fully reversible one-electron reductions as well as two irreversible oxidation events, the former may be of interest in small-molecule n-type semiconductors.15,16 The lower reduction potential of 2b (∼90 mV vs.2a) suggests that the reduced HOMO–LUMO gap is primarily due to a stabilization of the LUMO energy. Having the external thienyl substituents and the bulky Mes* protecting group, we rationalized an oxidative radical polymerization pathway occurring exclusively through the external α-positions.17,18 Though not as versatile as chemical polymerization methods, electropolymerization is still widely used,19–21 and offers several advantages over homogeneous methods such as the possibility to avoid side reactions with chemical reagents, control on polymer-growth rate via applied potential, as well as a high reproducibility. Electropolymerization of 2a and 2b was thus performed on fluorine doped tin oxide (FTO) glass substrates (Fig. S3, ESI†).22 Formation of films was readily visible after a few scans, and reversible colour changes from yellow-light green (neutral) to dark blue (oxidized) were evident (vide infra). The radical cations 2a˙+ and 2b˙+ were optimized and the Mulliken spin density is found to be delocalized over the dithienyl-fluorenylidene π-backbone with largest contributions at the external α-carbon of the thiophene rings (Fig. S4, ESI†). Noteworthy, no spin density is located on the carbons bonding to the pnictogen or the pnictogen itself. This is in agreement with a recent EPR study on a similarly delocalized phosphaalkene radical cation system.23 Very importantly, attempts to electropolymerize 1a and 1b were unsuccessful, confirming the typical polymerization via the, more accessible, external α-positions on the thiophenes (Fig. S3, ESI†).
E) of 2a compared to 2b. The 31P-NMR spectra of 1a and 1b display low-field signals at 273.7 and 264.9 ppm, which are characteristic of non-polarized PC bonds and phosphaalkenes in delocalized π-conjugated systems. Structure analysis reveals typical PC and AsC bond lengths of 1.693(5) (1a), 1.677(5) (2a) and 1.807(3) Å (1b), respectively (Fig. S14, ESI†). The monomers have a coplanar fluoren-9-ylidene pnictogen backbone (the heteroatoms are only 0.061(1) (1a), 0.063(1) (1b) and −0.007(2) (2a) above/below the least squares plane of the fluorene core). The UV-Vis spectra of 1a and 1b show lowest-energy absorption bands at 371 and 398 nm, respectively (Fig. 2a). In comparison, the λmax of fluorene derivatives such as 2,7-di-bromo-9H-fluorene and 2,7-dibromo-9-methylene-9H-fluorene occurs at 311 and 312 nm. In comparison to all-carbon analogues, the introduction of the pnictinidene unit causes substantial changes in the optical spectrum as well as in the electronic structure of the system, as evidenced by the calculated excited-state transitions (see Fig. S1, ESI†). Dithienyl derivative 2a shows an additional shoulder at 399 nm (Fig. 2a) and a red-shifted broad band centred at 485 nm (λonset 552 nm).13 Similarly, 2b displays an absorption band at 409 nm and a low-energy band centred at around 530 nm (λonset 596 nm); the red-shifted features are due to extended conjugation on the π-backbone through thienyl-functionalization. The frontier molecular orbitals of 2a and 2b show the similarities between P and As (Fig. S2, ESI†), their HOMO corresponds to delocalized π-orbitals across the fluorenylidene-thienyl backbone, whereas their LUMO to fulvene-core π* orbitals, with a dominant contribution of the pnictogen–carbon double bond, which is more stabilized for 2b compared to 2a. The red shifts going from phosphinidene to arsinidene are mainly due to LUMO stabilization (as well as HOMO−1 destabilization). The HOMO−3 displays some lone pair contribution accessible for further functionalization (e.g. metal coordination, vide infra), which could be investigated in sensor devices.14 Electron density difference maps (EDDMs) from TD-DFT calculations provide insights on the difference in electron density for the low-energy transitions (Fig. 2a and Fig. S2, ESI†). The lowest-energy feature corresponds to a HOMO–LUMO transition with charge transfer character from the thienyl fluorene backbone to the hetero-fulvenoid core. The transitions between 350 and 415 nm are mainly based on π–π* transitions involving the fulvenoid pnictogen–carbon antibonding orbitals, which confirms that the phosphaalkene and arsaalkene are intrinsic to the molecular electronic structure of the systems. The electrochemical properties of the monomers were investigated using cyclic voltammetry (Fig. 2b). Both monomers showed fully reversible one-electron reductions as well as two irreversible oxidation events, the former may be of interest in small-molecule n-type semiconductors.15,16 The lower reduction potential of 2b (∼90 mV vs.2a) suggests that the reduced HOMO–LUMO gap is primarily due to a stabilization of the LUMO energy. Having the external thienyl substituents and the bulky Mes* protecting group, we rationalized an oxidative radical polymerization pathway occurring exclusively through the external α-positions.17,18 Though not as versatile as chemical polymerization methods, electropolymerization is still widely used,19–21 and offers several advantages over homogeneous methods such as the possibility to avoid side reactions with chemical reagents, control on polymer-growth rate via applied potential, as well as a high reproducibility. Electropolymerization of 2a and 2b was thus performed on fluorine doped tin oxide (FTO) glass substrates (Fig. S3, ESI†).22 Formation of films was readily visible after a few scans, and reversible colour changes from yellow-light green (neutral) to dark blue (oxidized) were evident (vide infra). The radical cations 2a˙+ and 2b˙+ were optimized and the Mulliken spin density is found to be delocalized over the dithienyl-fluorenylidene π-backbone with largest contributions at the external α-carbon of the thiophene rings (Fig. S4, ESI†). Noteworthy, no spin density is located on the carbons bonding to the pnictogen or the pnictogen itself. This is in agreement with a recent EPR study on a similarly delocalized phosphaalkene radical cation system.23 Very importantly, attempts to electropolymerize 1a and 1b were unsuccessful, confirming the typical polymerization via the, more accessible, external α-positions on the thiophenes (Fig. S3, ESI†).
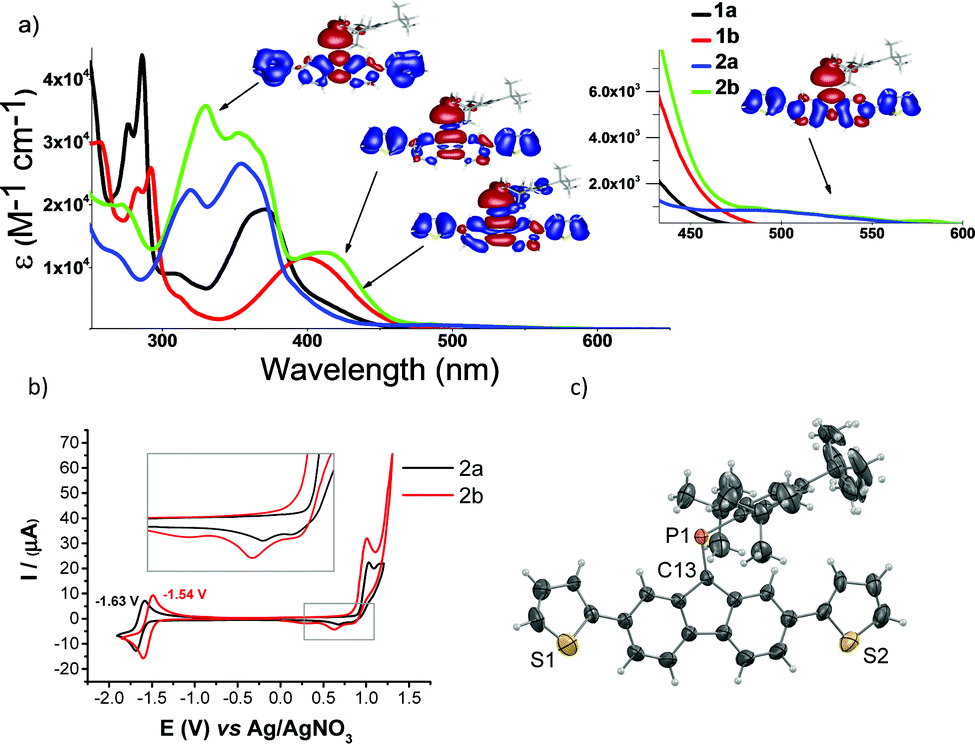 | ||
| Fig. 2 (a) UV-Vis absorption spectra of 1a-2b (DCM), inset: UV-Vis onsets; EDDMs selected transitions of 2b, depletion (blue) and increase (red) of electron density. See ESI† for details on computational methods. (b) Cyclic voltammograms of 2a and 2b (1 mM in DCM) vs. Ag/AgNO3 (10 mM in MeCN); glassy carbon electrode; scan rate: 100 mV s−1; (c) ORTEP representation of 2a; ellipsoids are drawn at 50% probability level. | ||
Energy dispersive X-ray spectroscopy (EDX) was used to confirm the presence of arsenic and phosphorus in all samples (Fig. S5, ESI†); the P/As![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S ratios were 1:2 in all appropriate samples. In order to corroborate a radical mechanism leaving the unsaturated pnictogen unit intact, the presence of PC and AsC moieties on the polymer films was confirmed through X-ray photoelectron spectroscopy (XPS).24 The analysis was performed by comparing the high-resolution spectra (C 1s, P 2p, As 3d, S 2p) of homogeneously deposited samples of monomers with polymer films (see Fig. 3c and Fig. S6, S7, ESI†). Based on the fully-characterized Mes*–EC fragments in the monomers and thus their chemical structure, standard procedures were used to characterize the XPS spectra (see ESI† for details).
:S ratios were 1:2 in all appropriate samples. In order to corroborate a radical mechanism leaving the unsaturated pnictogen unit intact, the presence of PC and AsC moieties on the polymer films was confirmed through X-ray photoelectron spectroscopy (XPS).24 The analysis was performed by comparing the high-resolution spectra (C 1s, P 2p, As 3d, S 2p) of homogeneously deposited samples of monomers with polymer films (see Fig. 3c and Fig. S6, S7, ESI†). Based on the fully-characterized Mes*–EC fragments in the monomers and thus their chemical structure, standard procedures were used to characterize the XPS spectra (see ESI† for details).
 | ||
| Fig. 3 Selected SEM pictures of poly-2b (a) poly-2b & FTO substrate, top view. (b) Cross-section picture of poly-2b (c) example of a high-resolution C 1s (left) and As 3d (right) spectra of 1b (top), 2b (middle), poly-2b (bottom); red curve: experimental, brown dotted curve: total fitted curve. Pictures of the respective films (right). (d) Selected orbital densities and energies (HOMO−3 to LUMO+3) for 2b, 2b-dimer, 2b-trimer, and 2b-tetramer. Isosurface value: 0.02 a.u. | ||
The signal corresponding to carbon in an sp3 environment was used as an internal reference across all samples (285 eV, Table S1, ESI†).25 For 1a, 2a, and poly-2a, the BE of the phosphorus (P 2p) electrons in a phosphaalkene environment was found to be 129.98 ± 0.08 eV (n = 9). Analogously, for 1b, 2b, and poly-2b, the BE of the arsenic (As 3d) electrons in an arsaalkene environment was found to be 42.29 ± 0.05 eV (n = 8). In agreement with the electronic structure of divalent sp2-hybridized (CE–R) moieties, the phosphorus (P 2p) and arsenic (As 3d) photo-ejected electrons from unsaturated pnictogen–carbon bonded units have lower BEs in comparison to sp3-hybridized and/or oxidized pnictogen species, which would typically be found several eV at higher energies.26 The high-resolution spectroscopic data clearly demonstrates the presence of phosphorus–carbon and arsenic–carbon double bonds in the respective polymer films. With the exception of several Ultraviolet Photoelectron Spectroscopy experiments on EC containing systems,27,28 the characterization of phosphaalkenes and arsaalkenes by XPS is unprecedented and the presented work shows its versatility to characterize low-valent main group systems. Scanning-electron-microscopy (SEM) experiments were performed to probe the morphology of the polymer films (Fig. 3 and Fig. S8, ESI†). Both poly-2a and poly-2b films were found to be homogeneous, and from a cross-section image, a thickness of 2 μm was approximated. According to the structural information obtained by single crystal X-ray crystallography and the film morphology by SEM, the presence of bulky Mes* substituents does not negatively impact the solid state packing and homogeneity of the monomers and polymers, respectively; the former is seen by π–π interactions <3.8 Å in monomeric 2a (Fig. S15, ESI†). The electric-potential-responsive nature of the polymer films is presented as proof-of-principle application to corroborate their functionality. Spectroelectrochemical experiments in a pure electrolyte solution confirmed the electrochromism of poly-2a and poly-2b, with the appearance of absorptions at ca. 700 and >1100 nm for the oxidized polymer films (Fig. 4c and Fig. S9, ESI†), as the potential is cycled (0 to 1.3 V). The isosbestic-like point at ca. 509 nm for poly-2a (poly-2b: 496 nm) indicates the polymer film reversibly interconverts between a neutral and oxidized state. The electrochromic reversibility (Fig. S9, ESI†) of a sample of poly-2b was investigated using chronoamperometry; at 600 nm, a contrast ratio of 58% was maintained through several cycles (Fig. 4d). Interestingly, a dark blue oxidized species of poly-2b maintained its colour for several days under inert conditions with no applied bias (Fig. S10, ESI†), which points to a stable doped polymer species. Calculated UV-Vis-NIR spectra of oligomeric cation systems qualitatively point toward polaron-based absorptions in the NIR (Fig. 4c and Fig. S11, ESI†), similar to the spectroelectrochemistry results.
 | ||
| Fig. 4 (a) UV-Vis spectra of Au(I) complexes of 2a-AuCl and poly-2a-AuCl in comparison to 2a and poly-2a. (b) preparation of molecular gold complex 2a-Au (i) [AuCl(tht)], DCM, 2 hours r.t. 2a, (ii) [AuCl(tht)], CH3CN, 3 weeks r.t. (c) Spectroelectrochemistry of poly-2a; DCM solution (0.1 mM n-Bu4NPF6); 0 to 1.3 V; scan rate: 100 mV s−1; inset: differential plot (left) (d) electrochromic reversibility of a sample of poly-2b at different selected absorption wavelengths (0 to 1.3 V at a scan rate of 100 mV s−1). | ||
The accessibility of the pnictogen lone pair for further functionalization was confirmed by coordination of 2a and poly-2a to Au(I) salts, which caused red-shifts on the absorption spectrum of around 96 nm and 153 nm (Fig. 4a), respectively. The large s-character of the arsenic lone pair appears to make 2a and poly-2a less susceptible towards gold(I) coordination. The UV-Vis-NIR spectra of solid poly-2a, poly-2a-AuCl, and poly-2b samples exhibit λonsets at 647, 800, and 737 nm (Fig. S12, ESI†), respectively; these are significantly red-shifted in comparison to monomer samples. As evidenced by DFT, the smaller HOMO–LUMO gap (e.g.2b: 2.78 eV, 2b-dimer: 2.37 eV, 2b-trimer: 2.28 eV, 2b-tetramer: 2.26 eV; see Table S4, ESI†) going from monomer to tetramer species is consistent with extended π-conjugation across the system (Fig. 3d and Fig. S13, ESI†); these fundamental gaps qualitatively agree with the optical gaps calculated from UV-Vis-NIR spectroscopy. The frontier orbital contribution of the introduced R–EC accepting units is substantial in the oligomeric systems, and these virtual orbitals effectively reach a band-like regime; for example, the LUMO to LUMO+1, LUMO+2 and LUMO+3 energy differences in the tetrameric model compound are less than 0.03, 0.06 and 0.1 eV, respectively.
In the search for π-conjugated materials incorporating heavier main group elements, we reported the synthesis and characterization of novel functionalized phosphaalkene (PC) (poly-2a) and arsaalkene (AsC) (poly-2b) π-conjugated polymers. To the best of our knowledge, this is the first example of a polymer containing AsC double bonds. The obtained units contain planar EC-fluorenylidene backbones. The polymer films display reversible electrochromic behavior from light yellow-green in the neutral state to deep blue in the oxidized state; additionally, the accessibility of a lone pair was confirmed by post-functionalization of 2a and poly-2a with Au(I) ions, which significantly perturbed the optoelectronics of the systems. These features demonstrate the versatility of these units for potential use in π-conjugated materials applications. Our current efforts focus on the soluble polymer version and profound photo-physical studies of these building blocks.
Support was provided by Lars-Hiertas memorial fund, the Olle-Engkvist foundation and the Vetenskapsrådet (Swedish research council 2013-4763), and the European COST network on Smart Inorganic Polymers (SIPs, CM1302). The authors thank Jacinto Sa for comments on the XPS analysis and Arvind Gupta for growing a crystal of 2a and solving the structure of 2a-AuCl. This paper is dedicated to Prof. Dr. Evamarie Hey-Hawkins on occasion of her 60th birthday.
Notes and references
- S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC.
- M. A. Shameem and A. Orthaber, Chem. – Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed.
- M. Stolar, J. Borau-Garcia, M. Toonen and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 3366–3371 CrossRef CAS PubMed.
- J. P. Green, Y. Han, R. Kilmurray, M. A. McLachlan, T. D. Anthopoulos and M. Heeney, Angew. Chem., Int. Ed., 2016, 55, 7148–7151 CrossRef CAS PubMed.
- B. L. Lucht and N. O. St Onge, Chem. Commun., 2000, 2097–2098 RSC.
- V. A. Wright and D. P. Gates, Angew. Chem., Int. Ed., 2002, 41, 2389–2392 CrossRef CAS PubMed.
- V. A. Wright, B. O. Patrick, C. Schneider and D. P. Gates, J. Am. Chem. Soc., 2006, 128, 8836–8844 CrossRef CAS PubMed.
- K. Naka, T. Umeyama and Y. Chujo, J. Am. Chem. Soc., 2002, 124, 6600–6603 CrossRef CAS PubMed.
- V. A. Wright and D. P. Gates, Angew. Chem., Int. Ed., 2002, 41, 2389–2392 CrossRef CAS PubMed.
- L. Weber, Chem. Ber., 1996, 129, 367–379 CrossRef CAS.
- H. Hopf, Angew. Chem., Int. Ed., 2012, 51, 11945–11947 CrossRef CAS PubMed.
- X. He and T. Baumgartner, RSC Adv., 2013, 3, 11334–11350 RSC.
- S. Rasmussen, in Encyclopedia of Polymeric Nanomaterials, ed. S. Kobayashi and K. Müllen, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 1–13 Search PubMed.
- A. Orthaber, H. Löfås, E. Öberg, A. Grigoriev, A. Wallner, S. H. M. Jafri, M.-P. Santoni, R. Ahuja, K. Leifer, H. Ottosson and S. Ott, Angew. Chem., Int. Ed., 2015, 54, 10634–10638 CrossRef CAS PubMed.
- A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1996, 8, 579–589 CrossRef CAS.
- Y. Dienes, M. Eggenstein, T. Kárpáti, T. C. Sutherland, L. Nyulászi and T. Baumgartner, Chem. – Eur. J., 2008, 14, 9878–9889 CrossRef CAS PubMed.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- J. Doskocz, M. Doskocz, S. Roszak, J. Soloducho and J. Leszczynski, J. Phys. Chem. A, 2006, 110, 13989–13994 CrossRef CAS PubMed.
- X.-M. Hu, Z. Salmi, M. Lillethorup, E. B. Pedersen, M. Robert, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Chem. Commun., 2016, 52, 5864–5867 RSC.
- H. H. Zhang, Y. N. Zhang, C. Gu and Y. G. Ma, Adv. Energy Mater., 2015, 5, 1402175 CrossRef.
- B.-B. Cui, J.-H. Tang, J. Yao and Y.-W. Zhong, Angew. Chem., Int. Ed., 2015, 54, 9192–9197 CrossRef CAS PubMed.
- T. A. Skotheim, Handbook of conducting polymers, CRC Press, 1997 Search PubMed.
- X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC.
- P. Pfluger and G. B. Street, J. Chem. Phys., 1984, 80, 544–553 CrossRef CAS.
- D. Briggs, Surface Analysis of Polymers by XPS and Static SIMS, Cambridge University Press, 1998 Search PubMed.
- S. Hoste, D. F. Van De Vondel and G. P. Van Der Kelen, J. Electron Spectrosc. Relat. Phenom., 1979, 17, 191–195 CrossRef CAS.
- V. Metail, A. Senio, L. Lassalle, J. C. Guillemin and G. Pfister-Guillouzo, Organometallics, 1995, 14, 4732–4735 CrossRef CAS.
- J. C. T. R. B.-S. Laurent, M. A. King, H. W. Kroto, J. F. Nixon and R. J. Suffolk, J. Chem. Soc., Dalton Trans., 1983, 755–759 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1431892, 1431893, 1485627 and 1486942. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08736a |
| This journal is © The Royal Society of Chemistry 2017 |