
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton-driven coordination-induced spin state switch (PD-CISSS) of iron(II) complexes†
René
Nowak
a,
Eko Adi
Prasetyanto
b,
Luisa
De Cola
b,
Beate
Bojer
c,
Renée
Siegel
c,
Jürgen
Senker
c,
Ernst
Rössler
d and
Birgit
Weber
 *a
*a
aAnorganische Chemie II, Universität Bayreuth, Universitätsstr. 30, NW I, 95440 Bayreuth, Germany. E-mail: weber@uni-bayreuth.de
bInstitut de Science et d'Ingenierie Supramoleculaire (ISIS), Université de Strasbourg, 67083 Strasbourg Cedex, France
cAnorganische Chemie III, Universität Bayreuth, 95440 Bayreuth, Germany
dExperimentalphysik II, Universität Bayreuth, 95440 Bayreuth, Germany
First published on 15th December 2016
Abstract
Here we report the completely reversible spin state switch of the naturally diamagnetic tris(bipyridine)iron(II) complex and the spin crossover complex bis(2,6-bis(1H-pyrazol-3-yl)pyridine)iron(II) by the variation of the pH followed by 1H-NMR, UV-Vis spectroscopy, and magnetic and relaxivity measurements in solution and as composite materials encapsulated in a zeolite matrix.
Spin crossover complexes are known for their ability to be switched between two different electronic states by a variety of physical or chemical stimuli; typical examples are temperature, pressure, light irradiation or host–guest interactions for porous materials.1 In the case of iron(II), this switching process occurs between a paramagnetic high spin (HS, S = 2) and a diamagnetic low spin (LS, S = 0) state. Based on this bistability, applications in the field of data storage,2 sensors3 or as responsive contrast agents4 were proposed. For applications as sensors, it would be interesting to switch the spin state reversibly as a function of pH. First iron(II) complexes were designed that can be switched upon pH change, however, so far only irreversibly.5 Here we show that it is possible to reversibly switch the magnetic properties of the naturally LS complex [Fe(bipy)3]2+ (1)6 and the SCO complex [Fe(bpp)2]2+ (2)7 (bipy = 2,2′-bipyridine; bpp = 2,6-bis(1H-pyrazol-3-yl)pyridine) upon protonation; both in solution and encapsulated in the zeolite NaY.
Investigations in solution of the spin crossover complex [Fe(bpp)2]2+ showed a SCO at around room temperature with T1/2 (where 50% of the iron centers are in the HS state) depending on the used solvent.7 The complex [Fe(bipy)3]2+ is a diamagnetic LS complex due to the strong ligand field of 2,2′-bipyridine. However, the protonation of a ligand, e.g. amino groups8a or the pyridine nitrogen, can influence the overall ligand field strength and by this the magnetic properties. Therefore, we started to investigate the influence of protonation on the spin state of this complex in solution. The 1H-NMR spectra of aqueous solutions (D2O) of [Fe(bipy)3]Cl2 (c = 0.007 mol L−1) at different pH values are given in Fig. 1A, which show at pH 6 the typical four signals for the protons of the [Fe(bipy)3]2+ ion (3(d), 4(t), 5(t), and 6(d)).9 The used labelling scheme is given in Fig. 1C.
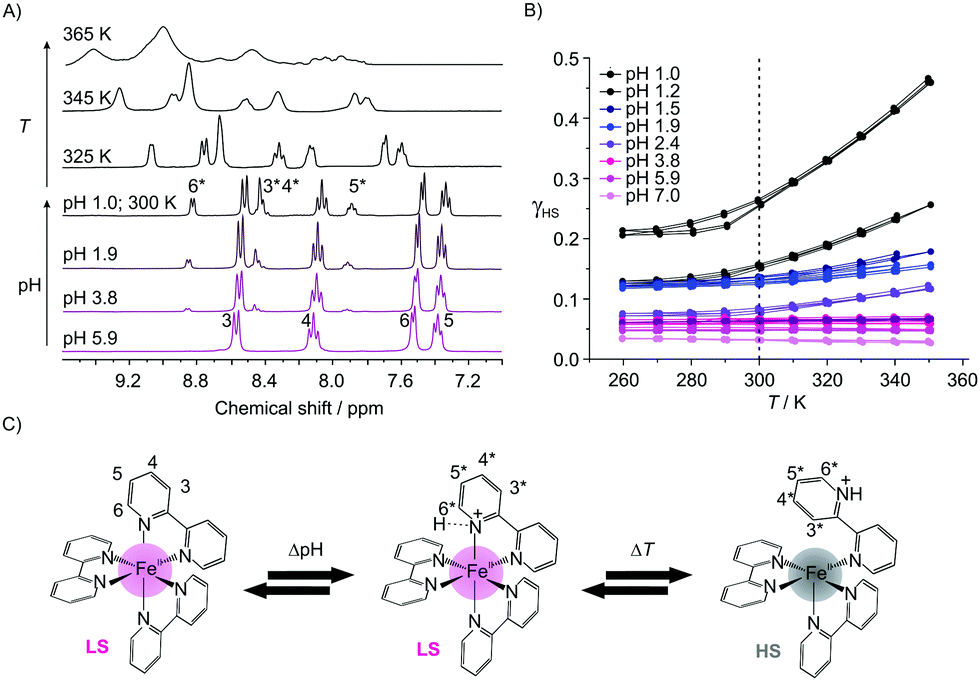 | ||
| Fig. 1 Characterization of [Fe(bipy)3]2+ in solution. (A) pH- and T-dependent 1H-NMR spectra of [Fe(bipy)3]Cl2 in D2O. (B) Plot of γHSversus T for the different solutions showing an increasing γHS due to a spin state change between LS and HS of the protonated complex. (C) Proposed mechanism for the interaction of [Fe(bipy)3]2+ with the protons with the labeling scheme used for the 1H-NMR spectra. | ||
When lowering the pH, new signals emerge in the aromatic region that are associated with the appearance of a diamagnetic protonated species [Fe(bipy)3H]3+ (3*(m), 4*(m), 5*(m), and 6*(d)). The relative integrals of these signals increase with proton concentration leading to a pKa value of 1.3 for this complex compared to a pKa value of 4.4 for free 2,2′-bipyridine.8b,c As shown in the ESI,† Fig. S7, the signals are shifted compared to the signals of the protonated free bipy ligand, thus complex dissociation can be ruled out. Upon heating of the solution at pH 1, a significant line broadening is observed, indicative of the formation of a paramagnetic species. This is confirmed by the T-dependent SQUID measurements of the solutions (H2O; c = 0.168 mol L−1) at different pH values. The results are given in Fig. 1B as a plot of the high spin fraction γHSvs. T. At pH 7 γHS is 0.03, which is almost negligible. When lowering the pH, γHS increases first slowly, and below pH 2.5 more abruptly with γHS values between 0.14 and 0.27 for pH 2 and 1. The solutions are now clearly paramagnetic. The absence of resonances of the paramagnetic species in the room temperature 1H NMR spectra in Fig. 1A can be explained with a significant line broadening. The observations point towards a pH-dependent spin state switch between a diamagnetic iron(II) LS species and a paramagnetic iron(II) HS species, starting at around pH 2.5. As already indicated by NMR spectroscopy, this spin state switch is temperature dependent. At pH 1 γHS varies between 0.21 and 0.46 while going from 260 K to 350 K. It is noteworthy that this pH- and T-dependent change is completely reversible and can be also followed via UV-Vis spectroscopy, as shown in the ESI,† Fig. S1. A loss of the charge transfer band at 520 nm is observed, which can be explained with a spin state change. This decrease is also observed when the temperature is increased at constant pH, in line with the SQUID and 1H-NMR data (ESI,† Fig. S1). Additionally, the π–π* transition at 300 nm is bathochromically shifted with the lowering of the pH, indicative of an interaction between the nitrogen donor of the pyridine rings and the protons. The 1H-NMR spectra and the results from the solution magnetic measurements suggest the formation of a protonated diamagnetic and a paramagnetic species.
In Fig. 1C, a mechanism is proposed to explain the experimental results. We suggest that at lower pH one of the pyridine nitrogens is protonated leading to a protonated diamagnetic species [Fe(bipy)3H]3+ that is observed in the 1H-NMR spectra. This protonation weakens the nitrogen–iron bond leading to a reversible bond break between the iron center and the protonated nitrogen of the bipy ligands, in line with a change of the coordination sphere and the spin state at the iron center. Then a proton-driven coordination-induced spin state switch (PD-CISSS) takes place. A change of the coordination sphere upon protonation was recently proposed for paramagnetic europium(III) complexes.10 This mechanism predicts the formation of a free coordination spot at the paramagnetic iron center that should increase dramatically the relaxivity of water. Consequently, aqueous solutions of 1 (c = 0.168 mol L−1) were characterized using field-cycling (FC) 1H-NMR relaxometry11 at different pH values. For comparison, aqueous solutions of the spin crossover complex 27 (c = 0.036 mol L−1) were investigated as well. The aqueous solution of the spin crossover complex is paramagnetic at room temperature (γHS ≅ 0.5). When the pH is lowered, the same mechanism as for 1 could be observed. Significant differences in the relaxivity values of 2 when the pH is lowered would support the proposed mechanism. In Fig. 2A the molar relaxivity Δr1 is plotted vs. the pH. The experiments yield 1H-relaxation of the water molecules that are influenced by the exchange at free coordination spots of the paramagnetic centers.11,12 For both complexes a strong pH-dependence of Δr1 is observed. At pH values below 4, a significant increase is observed in line with the proposed formation of a free site for the coordination of water in the inner coordination sphere of the iron as [Fe(bipy)3H(H2O)]3+. For 1 Δr1 is between 0.00(1) s−1 mmol−1 L (pH 7, LS, no coordination spot) and 0.18 s−1 mmol−1 L (pH 1, γHS = 0.27, free coordination spot) – corresponding to an increase of the molar relaxivity by a factor of 18.
 | ||
| Fig. 2 (A) Molar relaxivity Δr1(ν = 0.01 MHz) at room temperature plotted versus pH for 1 and 2. A steep increase of Δr1 is observed when the pH of the solution is lowered. The inset shows the observed relaxivity R1 plotted against the Larmor frequency ν. The RLM1 values at pH 2 and pH 7 are identical so the last one is omitted for clarity. (B) pH-Dependence of the room temperature γHS values calculated from SQUID data and γH+ values calculated from the 1H NMR spectra of 1. The arrows indicate the scale bar used. | ||
This increase is significantly higher than the one observed for switchable nickel(II) complexes with a factor of 3–7 (switching by light irradiation, the light-driven coordination-induced spin state switch LD-CISSS)13 or gadolinium-based systems with a factor of 3–5.14 For 2 the molar relaxivity varies between 0.06 s−1 mmol−1 L and 0.57 s−1 mmol−1 L corresponding to a factor of 9.5. The increase of the relaxivity of 2 starts at a slightly higher pH, indicating an influence of the used ligand.
In order to explore if the PD-CISSS can be also observed in composite materials, the iron(II) complexes were encapsulated in the supercages of zeolite faujasite NaY. The encapsulation was done using the impregnation method15 to avoid paramagnetic iron(II) high spin contamination of the sodalite cages. For the synthesis, NaY was used as a starting material, which has approx. 0.5 Brønsted acid sites per unit cell;16 their presence can be qualitatively verified by solid-state NMR spectroscopy (ESI,† Fig. S2). The samples 1@NaY and 2@NaY were fully characterized in the solid state by elemental analysis (C, H, N), AAS (Fe), TGA, solid-state NMR, UV-Visible and evanescent-wave IR spectroscopy. A detailed description of the characterization is given in the ESI.† Mössbauer spectroscopy was used to distinguish between the different possible iron sites in the zeolite cavity. The results confirm the formation of the complexes solely in the supercages, the absence of complexes on the surface (IR, UV-Vis, and Mössbauer), and were used to determine the sample composition (CHN, AAS, and Mössbauer). For the unit cell, the formula [Fe(bipy)3]0.4Na57H0.5[(AlO2)58(SiO2)134] was obtained for 1@NaY (0.13 wt% iron) and [Fe(bpp)2]0.8Na56H0.5[(AlO2)58(SiO2)134] for 2@NaY (0.36 wt% iron). This corresponds to 1 complex per 18 supercages (1@NaY) and 1 complex per 8 supercages (2@NaY). Please note that according to the room temperature Mössbauer spectra (ESI,† Fig. S5), the encapsulated spin crossover complex 2 is at room temperature, predominantly in the LS state. One unit cell (8 supercages) can absorb up to 260 water molecules.17 In combination with the Brønsted acid sites the zeolite cavities can be considered to be acidic with the pH depending strongly on the water contents (ESI,† Fig. S2). Removal of water, e.g. upon heating, will lower the pH and can be used to investigate the influence on the spin state of the metal center. Upon heating of the neat composite materials, for both samples, a completely reversible colour change from red to colourless (1@NaY), and yellow to colourless (2@NaY), is observed (Fig. 3 and ESI,† Fig. S3, Movie M1), which can be also followed by UV-Visible spectroscopy (loss of the MLCT-band from red/yellow to colourless). This colour change is linked to the loss of water in the cavities of the zeolite as derived from TG analysis, and the red and yellow colours are restored when 1@NaY and 2@NaY are treated with a drop of water or equilibrated in air for some time. Remarkably, the properties of 1 and 2 in the supercage of the zeolite are very similar although 2 is well known for its SCO properties while 1 is a stable LS complex. No indication for a colour change upon cooling is observed in both cases. This indicates that the magnetic properties of 2 are significantly influenced by the zeolite environment, in agreement with some preliminary results on encapsulated cobalt(II)18 and iron(II)19 complexes. The expected spin state change was analyzed using magnetic measurements in the 150–400 K temperature range. The results are given on the right side of Fig. 3 for 1@NaY (top) and 2@NaY (bottom). The room temperature magnetic susceptibility χmass of both samples is highly negative (−6 × 10−3 cm3 g−1 at 300 K each), characteristic for diamagnetic zeolite samples. Upon cooling and heating between RT and 150 K, no significant changes are observed. Subsequent heating of both samples to 400 K reveals a significant increase in the mass susceptibility in both cases, with a χmass value at 300 K of −3 × 10−3 cm3 g−1 for 1@NaY and 3 × 10−3 cm3 g−1 for 2@NaY. This corresponds to a change of the mass susceptibility (Δχmass) at room temperature of 3 × 10−3 cm3 g−1 for 1@NaY and 9 × 10−3 cm3 g−1 for 2@NaY. In the latter case the increase of χmass is more pronounced in line with the higher iron contents (three-fold). From this we conclude the generation of a paramagnetic complex inside the composite materials following the same mechanism as proposed for the solution samples in Fig. 1C. Upon heating, water in the zeolite is removed thus the pH inside the cavities is lowered leading to a protonation of one of the nitrogen donor atoms. In the ESI,† at the top of Fig. S9, the proposed mechanism inside the zeolite supercages is given. Please note that this spin state change and the associated colour change can be also observed for suspensions of the composite material 1@NaY in water, when the pH is lowered in small steps by the use of hydrochloric acid. A picture of this series where the pH was varied between 6.0 and 2.0 is given at the bottom of Fig. S9 (ESI†) together with the proposed mechanism. In line with the proposed coordination change at the iron center, a significant T1 shortage of the interzeolitic water is found for the solid composite material 2 between the wet (water vapour saturation) (27.79 ms) and dry (heated at 100 °C) (11.06 ms) states using solid-state NMR (see ESI,† Fig. S8). The composite material 1 was saturated with 0.1 M HCl and then heated in a solid-state NMR experiment; T1 is reduced gradually from 26.66 ms at 300 K, 19.38 ms at 320 K and 15.73 ms at 340 K to 11.93 ms at 375 K (see the ESI,† Fig. S8). We can conclude that inside the zeolite a proton-driven coordination-induced spin state switch (PD-CISSS) takes place.
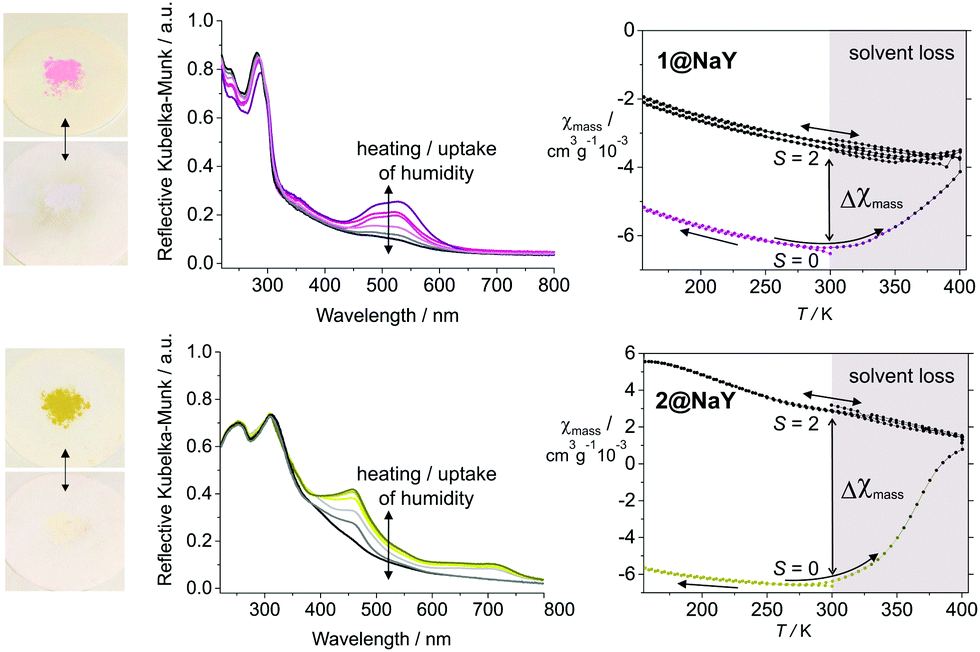 | ||
| Fig. 3 Change of the optical and magnetic properties of the samples 1@NaY (top) and 2@NaY (bottom) upon heating (solvent loss). On the left the reversible color change from red (1@NaY, top) and yellow (2@NaY, bottom) to colorless is illustrated showing the samples and the corresponding UV-Vis spectra where a loss/gain of the MLCT band is observed upon heating/standing on humid air. On the top right, the increase of the mass magnetic susceptibility (Δχmass) of 1@NaY is given. This increase depends on the iron contents of the sample and is higher for sample 2@NaY with the higher iron contents. The change of χmass is irreversible as the instrument operates under vacuum. | ||
In this manuscript we presented the pH-dependent magnetic properties of the complexes [Fe(bipy)3]2+ (1) and [Fe(bpp)2]2+ (2), both in solution and as composite materials encapsulated in the supercages of zeolite faujasite NaY. Based on the investigations in solution using 1H-NMR spectroscopy, magnetic measurements, UV-Vis spectroscopy and molar relaxivity measurements, a proton-driven coordination-induced spin state switch (PD-CISSS) was identified as the underlying mechanism. Due to the formation of a free coordination site upon protonation, the presented systems are promising candidates for pH-responsive contrast agents in MRI, provided that similar results can be obtained for nano-sized zeolite particle suspensions with high complex loading. The composite materials prepared so far show excellent long-term stability. One can see easily that the PD-CISSS and the resulting pH-dependent relaxation are linked to the used ligand type and are crucially influenced by the molecular structure and the mechanic-geometrical properties of the ligand. For the composite materials the zeolite environment can be further used to optimize the relaxivity with regard to the absolute values and the difference between the LS and the HS state. The presented hybrid materials are highly interesting for use as smart contrast agents in precise tumor detection as tumor tissues are known to exhibit a higher temperature and a lower pH (Warburg Effect,20 up to two decades21) compared to the surrounding tissue.
Financial support of the SFB 840 and from the University of Bayreuth is gratefully acknowledged. R. N. was supported by a Chemiefonds fellowship, an ERASMUS scholarship and the ENB Elite Network of Bavaria.
Notes and references
- (a) Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013 Search PubMed; (b) S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- O. Kahn, Science, 1998, 279, 44–48 CrossRef CAS.
- (a) G. J. Halder, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed; (b) E. Coronado, M. Giménez-Marqués, G. Mínguez Espallargas, F. Rey and I. J. Vitórica-Yrezábal, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS PubMed.
- (a) R. N. Müller, L. Vander Elst and S. Laurent, J. Am. Chem. Soc., 2003, 125, 8405–8407 CrossRef PubMed; (b) C. Rajadurai, M. Ruben and D. Kruk, EP2072062B1, 2012; (c) V. Stavila, M. Allali, L. Canaple, Y. Stortz, C. Franc, P. Maurin, O. Beuf, O. Dufay, J. Samarut, M. Janier and J. Hasserodt, New J. Chem., 2008, 32, 428 RSC.
- J. Hasserodt, J. L. Kolanowski and F. Touti, Angew. Chem., Int. Ed., 2014, 53, 60–73 CrossRef CAS PubMed.
- F. Basolo, J. C. Hayes and H. M. Neumann, J. Am. Chem. Soc., 1954, 76, 3807–3809 CrossRef CAS.
- S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2011, 40, 12021–12024 RSC.
- (a) Y.-H. Luo, M. Nihei, G.-J. Wen, B.-W. Sun and H. Oshio, Inorg. Chem., 2016, 55, 8147–8152 CrossRef CAS PubMed; (b) C. V. Krishnan, C. Creutz, H. A. Schwarz and N. Sutin, J. Am. Chem. Soc., 1983, 105, 5617–5623 CrossRef CAS; (c) NIST Database, acid dissociation constants.
- L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szłyk, Magn. Reson. Chem., 2010, 48, 450–457 CrossRef CAS PubMed.
- D. Sannasy, H. M. Marques, M. A. Fernandes and A. S. de Sousa, Chem. Commun., 2014, 50, 1582–1584 RSC.
- D. Kruk, A. Herrmann and E. A. Rössler, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 63, 33–64 CrossRef CAS PubMed.
- (a) E. Tóth, R. D. Bolskar, A. Borel, G. González, L. Helm, A. E. Merbach, B. Sitharaman and L. J. Wilson, J. Am. Chem. Soc., 2005, 127, 799–805 CrossRef PubMed; (b) M. P. Lowe, D. Parker, O. Reany, S. Aime, M. Botta, G. Castellano, E. Gianolio and R. Pagliarin, J. Am. Chem. Soc., 2001, 123, 7601–7609 CrossRef CAS PubMed.
- (a) M. Dommaschk, M. Peters, F. Gutzeit, C. Schütt, C. Näther, F. D. Sönnichsen, S. Tiwari, C. Riedel, S. Boretius and R. Herges, J. Am. Chem. Soc., 2015, 137, 7552–7555 CrossRef CAS PubMed; (b) S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sonnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed.
- (a) W. Zhang, J. A. Peters, F. Mayer, L. Helm and K. Djanashvili, J. Phys. Chem. C, 2015, 119, 5080–5089 CrossRef CAS; (b) C.-Y. Cao, Y.-Y. Shen, J.-D. Wang, L. Li and G.-L. Liang, Sci. Rep., 2013, 3, 1024 Search PubMed.
- W. H. Quayle, G. Peeters, G. L. De Roy, E. F. Vansant and J. H. Lunsford, Inorg. Chem., 1982, 21, 2226–2231 CrossRef CAS.
- (a) K. J. Thomas and V. Ramamurthy, Langmuir, 1998, 14, 6687–6692 CrossRef CAS; (b) V. Jayathirtha Rao, D. L. Perlstein, R. J. Robbins, P. H. Lakshminarasimhan, V. Ramamurthy, H.-M. Kao and C. P. Grey, Chem. Commun., 1998, 269–270 RSC.
- R. F. Howe, in Tailored Metal Catalysts, ed. Y. Iwasawa, Springer Netherlands, Dordrecht, 1986, pp. 141–182 Search PubMed.
- (a) K. Mizuno and J. H. Lunsford, Inorg. Chem., 1983, 22, 3484–3486 CrossRef CAS; (b) S. K. Tiwary and S. Vasudevan, Chem. Phys. Lett., 1997, 277, 84–88 CrossRef CAS; (c) S. K. Tiwary and S. Vasudevan, Inorg. Chem., 1998, 37, 5239–5246 CrossRef CAS.
- (a) Y. Umemura, Y. Minai, N. Koga and T. Tominaga, Chem. Commun., 1994, 893 RSC; (b) R. Vijayalakshmi, S. M. Yusuf and S. K. Kulshreshtha, J. Phys. Chem. Solids, 2004, 65, 975–979 CrossRef CAS.
- O. Warburg, Naturwissenschaften, 1924, 12, 1131–1137 CrossRef CAS.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the methods used and a complete description of the synthesis of the samples and their characterization. See DOI: 10.1039/c6cc08618g |
| This journal is © The Royal Society of Chemistry 2017 |