
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hybrid carbocyclic/N-heterocyclic carbene ligand†
Christian
Jandl
and
Alexander
Pöthig
 *
*
Catalysis Research Center & Department of Chemistry, Technische Universität München, Ernst-Otto-Fischer-Str. 1, 85747 Garching, Germany. E-mail: alexander.poethig@tum.de
First published on 8th December 2016
Abstract
We report the synthesis and characterisation of the first complex bearing a bidentate carbocyclic/N-heterocyclic carbene ligand, which represents an unprecedented combination of these two carbene classes. Starting from an unsubstituted cycloheptatrienylidene–Pd complex, the ligand was built up in an on-site synthesis at the metal center. DFT calculations were employed to understand the crucial allyl-to-alkyl rearrangement of the carbocycle in this sequence and all its binding modes during the reaction cascade were crystallographically characterised.
While N-heterocyclic carbenes (NHCs) have established themselves as a valuable and versatile ligand class in the toolkit of organometallic chemistry,1–5 their carbocyclic relatives have only rarely made it into the focus of attention yet. Same as for the NHCs,6,7 the history of carbocyclic carbenes begins in 1968 when the first cyclopropenylidene complex was discovered by Öfele.8 A second major class of carbocyclic carbenes is represented by the cycloheptatrienylidenes (CHTs), the first examples of which were reported in 1978 by Jones et al.9 In 2006 Bertrand et al. succeeded in the isolation of the first free cyclopropenylidene derivative,10 but in contrast to the isolation of the first free NHC by Arduengo in 199111 this did not prompt a comparable breakthrough, although some studies suggested that carbocyclic carbene ligands have similar potential in catalysis.12–14 The lag in their development also shows up in the fact that only within the last two years the first Au–CHT complex15 and a Cu-based transmetallation agent for cyclopropenylidene ligands16 were reported – landmarks long established in NHC chemistry.3
Like the NHCs, CHTs and cyclopropenylidenes act mainly as σ-donors with only minor backbonding and all three classes generally feature similar M–Ccarbene bond lengths.12 A crucial difference from NHCs is the reactivity of the carbene-C in CHT complexes towards nucleophiles. On the one hand, this may limit their applicability as steering ligands, since the carbene–metal bond can be cleaved during catalytic transformations. On the other hand, a ligand centred reactivity can be utilised as has recently been shown by Iluc et al. for a Pd complex bearing a reactive non-cyclic carbene ligand which is capable of C–H and related bond activations.17,18 The reactivity of CHT ligands also opens up the opportunity to use them as starting materials for an on-site build-up of functionalised cycloheptatriene-based ligand systems.19,20 On this basis we present a chelating ligand system, which combines a stable and inert NHC donor group with a more flexible and reactive CHT moiety (see Scheme 1).
 | ||
| Scheme 1 Left: General structure of an N-heterocyclic (NHC) and a carbocyclic carbene ligand (CHT). Right: Chelating hybrid CHT/NHC-ligand (this work). | ||
Starting from the imidazolium-substituted η3-cycloheptatrienide complex 1, previously reported by us and easily accessible from [PdBr2(CHT)]2,20 the imidazolium moiety is deprotonated using caesium carbonate (see Scheme 2). The so-formed NHC intramolecularly coordinates to Pd, upon which a rearrangement of the allyl(η3)-bound seven-membered ring to an alkyl(η1) binding mode takes place to form the monomeric complex 2. As a by-product of this synthesis we could isolate and characterise complex 3 in which the allyl binding mode of the seven-membered ring is maintained (see 1.4 in the ESI†). The coordinating acetonitrile in complex 2 is easily removed to yield the acetonitrile-free complex 4 upon drying under vacuum. To saturate the valence of Pd(II) in 4 we assume the formation of different bromide-bridged oligomers in the solid state (see Scheme S1 in the ESI†). Such oligomers possibly also exist as secondary species in an equilibrium in solution as has recently been shown for a related compound.21 This might explain why 2 and 4 were unsuitable for growing single crystals. Therefore, we tried to exchange the bromido ligand by precipitation with silver salts. The treatment of 2 with silver acetate yields the dimeric complex 5, in which both the bromido and acetonitrile ligands are exchanged for bridging acetate. The crystal structure of the resulting complex (Fig. 2, left) is discussed below. Using silver tetrafluoroborate the monomeric complex 6 bearing two acetonitrile ligands is obtained.
 | ||
| Scheme 2 Synthesis of cycloheptatrienyl/NHC complex 2 (including byproduct 3) and subsequent removal of acetonitrile in vacuo as well as its reaction with silver salts leading to ligand substitution. | ||
In the 13C NMR spectra the carbene resonances of the NHCs are found at 173.88 ppm (2), 172.68 ppm (5) and 167.99 ppm (6), which correspond to the donor strength of the respective trans-located ligand.22 In the 1H NMR the alkyl–H signal of the cycloheptatrienyl-moiety appears at 3.17 ppm for 2 and at 3.24 ppm for 6. These signals belong to the monomeric species as shown in Scheme 2, as 6 lacks anions able to bridge and MS data confirm a coordinating acetonitrile molecule for 2. For 5, however, two species are present in CD3CN: one with a comparable shift of 3.24 ppm and another with a significant highfield shift to 2.61 ppm. By contrast, in non-coordinating CD2Cl2 only one species is observed with an alkyl–H signal at 2.63 ppm. The latter belongs to the dimeric species also found in the solid state structure (vide infra), the crystals for which were grown from non-coordinating solvents. In a coordinating solvent like acetonitrile, these dimers apparently are partly cleaved thus forming monomers exhibiting a downfield shift of the alkyl–H signal similar to monomeric 2 and 6.
For a deeper understanding of the reaction pathway, we performed DFT calculations of the essential allyl-to-alkyl rearrangement. Starting from deprotonated 1 with the central allylic carbon atom in β-position to the substituent (1NHC-β), the free energy profile of the reaction in acetonitrile solution is depicted in Fig. 1. In analogy to the established dynamic behaviour of η3-cycloheptatrienide complexes,20 the Pd atom first moves along the ring from the β- to the α-position (1NHC-α). In the next step, the coordination of the NHC to Pd prompts the actual rearrangement: the seven-membered ring bends from a nearly planar conformation to a boat-shape with a tetrahedral alkyl-carbon (2Br). All activation barriers are quite low indicating a very fast reaction (for more details see the ESI†).
 | ||
| Fig. 1 Simplified Gibbs free energy profile of the rearrangement step of the reaction from deprotonated 1 to 2 in acetonitrile according to DFT calculations (for complete data see the ESI†). | ||
The calculated geometry is in good accordance with the experimentally derived solid state structure of 5 (Fig. 2, left). Of such η1-bound cycloheptatrienyl complexes only three examples in transition metal chemistry and some more involving tin have been crystallographically characterised so far.23–30 A η1-cycloheptatrienyl complex has also been proposed as a short-lived intermediate in the reaction of CHT complexes with nucleophiles.20 In agreement with previous reports, the seven-membered ring adopts a distinct boat-conformation as required by the tetrahedral environment of the alkyl-bound carbons C1 and C23, which feature bond lengths of Pd1–C1 = 2.023(7) Å, C1–C2 = 1.482(15) Å, C1–C7 = 1.496(12) Å, Pd2–C23 = 2.008(9) Å, C23–C24 = 1.500(14) Å and C23–C29 = 1.489(12) Å. The carbene–metal bond lengths of the NHCs are Pd1–C8 = 1.968(9) Å and Pd2–C30 = 1.947(8) Å. The bridging acetate ligands create a Pd–Pd interaction (Pd1–Pd2 = 2.9147(12) Å), which arranges the cycloheptatrienyl/NHC ligands in a stacked fashion locking the alkyl–protons in between the two ligands, which explains the highfield NMR shift observed for 5 as mentioned before.
 | ||
| Fig. 2 Molecular structures of 5 (one molecule out of four in the asymmetric unit), 7 and 8 in the solid state with ellipsoids at a probability level of 30% for 5 and 50% for 7 and 8. DiPP-groups and acetate ligands are simplified as wireframes where necessary and hydrogen atoms as well as co-crystallised solvent molecules are omitted for clarity. Selected distances (Å) and angles (°). 5: Pd1–Pd2 2.9147(12), Pd1–C1 2.023(7), Pd1–C8 1.968(9), Pd2–C23 2.008(9), Pd2–C30 1.947(8), C1–C2 1.482(15), C1–C7 1.496(12), C23–C24 1.500(14), C23–C29 1.489(12), C1–Pd1–C8 81.6(4), C23–Pd2–C30 81.7(4). 7: Pd1–Br1 2.4915(7), Pd1–Br1i 2.5120(8), Pd1–C1 2.005(4), Pd1–C8 1.957(5), C1–C2 1.397(6), C1–C7 1.411(7), Br1–Pd1–Br1i 83.43(2), C1–Pd1–C8 79.71(18), C1–C7–N1–C8 1.7(6). 8: Pd1–Br1 2.4859(4), Pd1–Br2 2.4941(4), Pd1–C1 1.994(3), Pd1–C8 1.967(2), C1–C2 1.403(3), C1–C7 1.422(3), Br1–Pd1–Br2 89.66(1), C1–Pd1–C8 80.60(10), Br2–Pd1–C8 164.56(7), C1–C7–N1–C8 −0.9(3). | ||
Having established the chelating η1-cycloheptatrienyl/NHC ligand, the next step to create the envisioned neutral hybrid ligand consists of a hydride abstraction from the cycloheptatrienyl-moiety of 4. In analogy to the original protocols of Jones et al.9 this was performed via the reaction with tritylium tetrafluoroborate as shown in Scheme 3. The obtained dimeric CHT/NHC complex 7 can be treated with tetrabutylammonium bromide to precipitate the neutral monomeric complex 8.
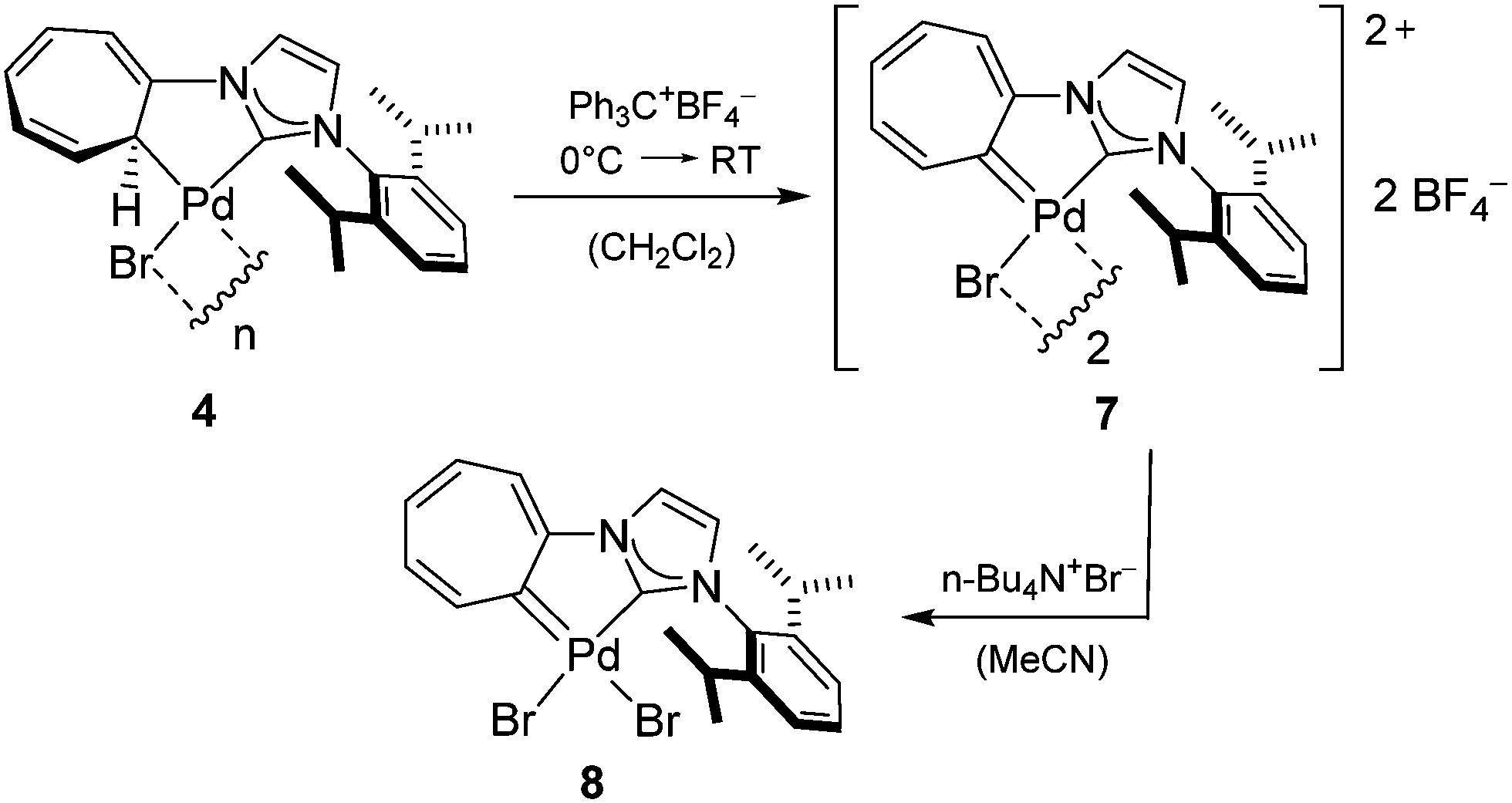 | ||
| Scheme 3 Synthesis of CHT/NHC complex 7 from 4 and its reaction with tetrabutylammonium bromide to monomeric complex 8. | ||
The chemical shift of the carbocyclic carbene-C appears considerably more downfield than that of the NHC: 185.16 ppm vs. 169.59 ppm for 7 (in CD3CN) and 191.3 ppm vs. 170.8 ppm for 8 (in the solid state). However, this is still a tremendous upfield-shift compared to literature-known Pd complexes of isolated CHT-ligands, for which carbene resonances have been reported in a region of ca. 205–230 ppm.12,19,20 For instance, the carbene resonance of [PdBr2(CHT)]2 appears at 209.1 ppm in the solid state. This difference can be attributed to an electronic interaction between the CHT- and NHC-moieties so that the donor function of the N-atoms extends to the carbocyclic carbene.
Single crystal X-ray diffraction confirms the structures of 7 (Fig. 2, middle) and 8 (Fig. 2, right). The CHT and NHC-moieties are coplanar (with the seven-membered ring being only slightly bent) supporting the idea of an aromatic system that allows electronic communication. The Pd atom is located slightly out of the C1–C7–N1–C8 plane in both complexes. Also in both cases the Pd–Ccarbene bond of the carbocyclic carbene is significantly longer than that of NHC (2.005(4) Å vs. 1.957(5) Å for 7, 1.994(3) Å vs. 1.967(2) Å for 8) and of literature-known CHT–Pd complexes.12,19,20 This agrees well with the aforementioned electron-donation from the NHC to the CHT which then weakens the Pd–CHT π-backbond. There is also a steric interaction between the H-atom on C2 and one bromide ligand, which forces a significant distortion of the square-planar coordination geometry in 8. Using the concept introduced by Houser et al.31 and improved by Kubiak et al.32 a value of 0.143 can be calculated for the τδ parameter (where τδ = 0 represents an ideal square planar and τδ = 1 an ideal tetrahedral coordination). The effect is also present in 7, but less pronounced (τδ = 0.042). In this context it is not surprising that the mentioned H-atom is also marked by an unusual downfield shift in the 1H NMR spectra as compared to unsubstituted CHT–Pd complexes (10.73 ppm in CD3CN for 7 and 10.32 ppm in DMSO-d6 for 8).12,19,20
With this effect in mind the Pd–Br bond lengths may no longer be an ideal indicator for the binding properties of the trans-located carbene ligands, but nevertheless it is worthwhile using this chance for a direct comparison between these two carbene donor moieties. It can be seen that the bromide trans to the carbocyclic carbene features the longer Pd–Br bond length in both cases (2.5120(8) Å vs. 2.4815(7) Å for 7, 2.4941(4) Å vs. 2.4859(4) Å for 8), which is compatible with our expectation that the CHT ligand exhibits a stronger trans-influence than the NHC ligand. This is in agreement with the very few examples of NHC complexes for which also an exact analogue bearing a CHT has been reported suggesting that this observation can in fact be generalised.13,33,34
Another interesting feature is the orientation of the carbene–metal bonds in relation to the CHT and NHC rings, because the formation of the five-membered metallacycle in 7 and 8 requires some deviation from the ideal binding geometries. To describe such distortions the NHC tilting angle ΘNHC defined by Chaplin et al. as the angle between the metal, carbene-C and the centroid of the NHC can be used, where 180° corresponds to the ideal coordination geometry.35 Applying the same concept to the CHT ligand, ΘNHC and ΘCHT values of 167.70° vs. 175.67° for 7 and 163.53° vs. 174.85° for 8 are obtained. In both cases the distortion is mostly an in-plane displacement of the metal. The above values illustrate that the distortion affects the NHC a lot more than the CHT suggesting that the CHT–metal bond is more rigid and therefore less susceptible to deformations of the binding geometry in comparison to NHCs.
In conclusion, almost half a century after their common year of discovery, carbocyclic and N-heterocyclic carbenes have finally been combined in the ligand motif presented in this communication. During the on-site synthesis the seven-membered ring of the starting compound [PdBr2(CHT)]2 switches its binding mode several times: first, from a η1-carbene to a η3-allyl mode in 1, then to a η1-alkyl mode in 4 and eventually back to the η1-carbene mode in 7. Spectroscopic and crystallographic results highlight the influence of this unusual chelating carbene ligand on the coordination environment of Pd. More importantly, they indicate an electronic interaction between the directly connected CHT- and NHC-moieties. By comparison, most chelating bis-NHC ligands feature linkers which allow no electronic communication between the NHC moieties.36–38 Given the various catalytic applications of such bis-NHC complexes,36–38 the chelating system of CHT and NHC adds even more potential as it can profit not only from their electronic interaction, but also from the reactivity of the carbocyclic carbene-C. In our continuing research we now focus on tuning the CHT/NHC motif by ligand modifications and employing a broader range of metals to explore the potential of these unique mixed carbene ligands in catalysis and other applications.
This work is dedicated to Dr Karl Öfele for his pioneering work in carbene-chemistry. C. J. gratefully acknowledges support from the TUM Graduate School. We thank M. Bitzer and E. Hahn for measuring the ESI mass spectra and Dr G. Raudaschl-Sieber for recording the solid state NMR spectra. Prof. W. A. Herrmann and Prof. F. E. Kühn are acknowledged for their continuous support.
References
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed., 1997, 36, 2162–2187 CrossRef CAS.
- W. A. Herrmann, Angew. Chem., 2002, 114, 1342–1363 CrossRef.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., 2008, 120, 3166–3216 CrossRef.
- T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- H.-W. Wanzlick and H.-J. Schönherr, Angew. Chem., 1968, 80, 154 CrossRef.
- K. Öfele, J. Organomet. Chem., 1968, 12, P42 CrossRef.
- K. Öfele, Angew. Chem., 1968, 80, 1032–1033 CrossRef.
- N. T. Allison, Y. Kawada and W. M. Jones, J. Am. Chem. Soc., 1978, 100, 5224–5226 CrossRef CAS.
- V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2006, 312, 722–724 CrossRef CAS PubMed.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- K. Öfele, E. Tosh, C. Taubmann and W. A. Herrmann, Chem. Rev., 2009, 109, 3408–3444 CrossRef PubMed.
- W. A. Herrmann, K. Öfele, S. K. Schneider, E. Herdtweck and S. D. Hoffmann, Angew. Chem., Int. Ed., 2006, 45, 3859–3862 CrossRef CAS PubMed.
- Q. Yao, M. Zabawa, J. Woo and C. Zheng, J. Am. Chem. Soc., 2007, 129, 3088–3089 CrossRef CAS PubMed.
- R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2014, 53, 9369–9371 CrossRef CAS PubMed.
- Y. D. Bidal, M. Lesieur, M. Melaimi, D. B. Cordes, A. M. Z. Slawin, G. Bertrand and C. S. J. Cazin, Chem. Commun., 2015, 51, 4778–4781 RSC.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2015, 34, 4684–4692 CrossRef CAS.
- C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048–9051 RSC.
- K. Mantas-Öktem, K. Öfele, A. Pöthig, B. Bechlars, W. A. Herrmann and F. E. Kühn, Organometallics, 2012, 31, 8249–8256 CrossRef.
- C. Jandl, K. Öfele, F. E. Kühn, W. A. Herrmann and A. Pöthig, Organometallics, 2014, 33, 6398–6407 CrossRef CAS.
- C. Jandl, S. Stegbauer and A. Pöthig, Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 509–513 CAS.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- D. M. Heinekey and W. A. G. Graham, J. Organomet. Chem., 1982, 232, 335–343 CrossRef CAS.
- S. Nemeh, R. J. Flesher, K. Gierling, C. Maichle-Mössmer, H. A. Mayer and W. C. Kaska, Organometallics, 1998, 17, 2003–2008 CrossRef CAS.
- Y. W. Chan and K. S. Chan, Chem. Commun., 2011, 47, 4802–4804 RSC.
- J. E. Weidenborner, R. B. Larrabee and A. L. Bednowitz, J. Am. Chem. Soc., 1972, 94, 4140–4144 CrossRef CAS.
- L. K. K. LiShingMan, J. G. A. Reuvers, J. Takats and G. Deganello, Organometallics, 1983, 2, 28–39 CrossRef CAS.
- I. D. Gridnev and O. L. Tok, J. Am. Chem. Soc., 2003, 125, 14700–14701 CrossRef CAS PubMed.
- I. D. Gridnev and M. K. C. del Rosario, Organometallics, 2005, 24, 4519–4527 CrossRef CAS.
- W. Setaka, K. Hirai, H. Tomioka, K. Sakamoto and M. Kira, Chem. Commun., 2008, 6558–6560 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211–3217 CrossRef CAS PubMed.
- C. Taubmann, E. Tosh, K. Öfele, E. Herdtweck and W. A. Herrmann, J. Organomet. Chem., 2008, 693, 2231–2236 CrossRef CAS.
- K.-T. Chan, Y.-H. Tsai, W.-S. Lin, J.-R. Wu, S.-J. Chen, F.-X. Liao, C.-H. Hu and H. M. Lee, Organometallics, 2010, 29, 463–472 CrossRef CAS.
- J.-N. Luy, S. A. Hauser, A. B. Chaplin and R. Tonner, Organometallics, 2015, 34, 5099–5112 CrossRef CAS.
- E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS.
- J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic, computational and crystallographic detailss. CCDC 1510761–1510764. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08468k |
| This journal is © The Royal Society of Chemistry 2017 |