
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric rhenium tricarbonyl complexes show superior luminescence properties in live cell imaging†
Lukasz J.
Raszeja‡
a,
Daniel
Siegmund‡
a,
Anna L.
Cordes
a,
Jörn
Güldenhaupt
b,
Klaus
Gerwert
b,
Stephan
Hahn
c and
Nils
Metzler-Nolte
 *a
*a
aFaculty of Chemistry and Biochemistry, Inorganic Chemistry I – Bioinorganic Chemistry, Ruhr-University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: Nils.Metzler-Nolte@rub.de
bFaculty of Biology and Biotechnology, Chair of Biophysics, Ruhr-University Bochum, Universitätsstr. 150, 44801 Bochum, Germany
cRuhr-University Bochum, Zentrum für Klinische Forschung (ZKF), Universitätsstraße 150, 44780 Bochum, Germany
First published on 23rd December 2016
Abstract
The synthesis and photophysical properties of a novel series of rhenium tricarbonyl complexes based on tridentate phenanthridinyl-containing ligands are described. Photophysical data reveal beneficial luminescence behaviour especially for compounds with an asymmetric ligand set. These advantageous properties are not limited to organic solvents, but indeed also improved in aqueous solutions. The suitability of our new rhenium complexes as potent imaging agents has been confirmed by fluorescence microscopy on living cancer cells, which also confirms superior long-time stability under fluorescence microscopy conditions. Colocalisation studies with commercial organelle stains reveal an accumulation of the complexes in the endoplasmic reticulum for all tested cell lines.
A basic idea of medicinal bioinorganic chemistry is to get a deeper understanding of fundamental cellular processes. This includes also the quest for new insights into the modes of action of metal based drugs and the identification of their subcellular targets. For this purpose, fluorescence microscopy is an indispensable tool in order to verify the cellular distribution or accumulation of biologically active compounds. Although, it is common to use organic dyes for fluorescence microscopy investigations and biological assays, emissive metal complexes have gained an increasing interest in this field.1–7 Especially d6 low spin complexes of Ir(III), Ru(II) and Re(I) show highly promising photophysical properties. In general, these complexes benefit from large Stokes shifts, long emission life times and enhanced photostability.8,9 Furthermore, their photophysical behaviour can be tuned by the modification and variation of their ligands. Published emissive Re(I) complexes are frequently based on the fac-Re(CO)3 core where bipyridine or phenanthroline derivatives10–19 or tridentate chelating ligands20–27 occupy the remaining coordination sites.
Herein we report on the favourable photophysical properties of novel rhenium tricarbonyl complexes, which are based on phenanthridinyl(phen)-containing tridentate ligands. Most importantly, these complexes exhibit their beneficial luminescence behaviour not only in degassed acetonitrile but also in air-equilibrated water. Further, these advantageous luminescence properties are underlined by the successful application of the described complexes as imaging agents in live cell fluorescence microscopy.
The ligands designed for our studies unite two important characteristics for potential imaging applications. Their phen-based extended π-system ensures the availability of electronically accessible ligand orbitals for MLCT or IL transitions which are crucial for the luminescence behaviour of the resulting complexes. Secondly, being benzoic acid derivatives, ester hydrolysis and subsequent conjugation to a variety of biologically active molecules or drugs enables the usage of these complexes as luminescent tags for tracing such bioactive compounds.28 To tune and evaluate the influence of phen on the photophysical properties, one of the phen moieties in the symmetrical ligand 1 is replaced by a well-known picolyl or quinolyl moiety respectively, leading to unsymmetrical ligands 2 and 3. Symmetric ligand 1 was prepared in one step by the reaction of 6-(chloromethyl)phenanthridine with 4-(aminomethyl)benzoic acid methyl ester.28 The asymmetric ligands 2 and 3 were synthesized through the sequential reaction of 4-(aminomethyl)benzoic acid methyl ester with 2-picolyl chloride or 2-(chloromethyl)quinoline followed by the reaction with 6-(chloromethyl)phenanthridine. Finally, the ligands were reacted with the [Re(CO)3(H2O)3]Br precursor under microwave conditions to yield the desired rhenium tricarbonyl complexes 4–6 (Fig. 1). Synthetic details and full characterization of the compounds, including single crystal X-ray structures for 4–6 can be found in the ESI.†
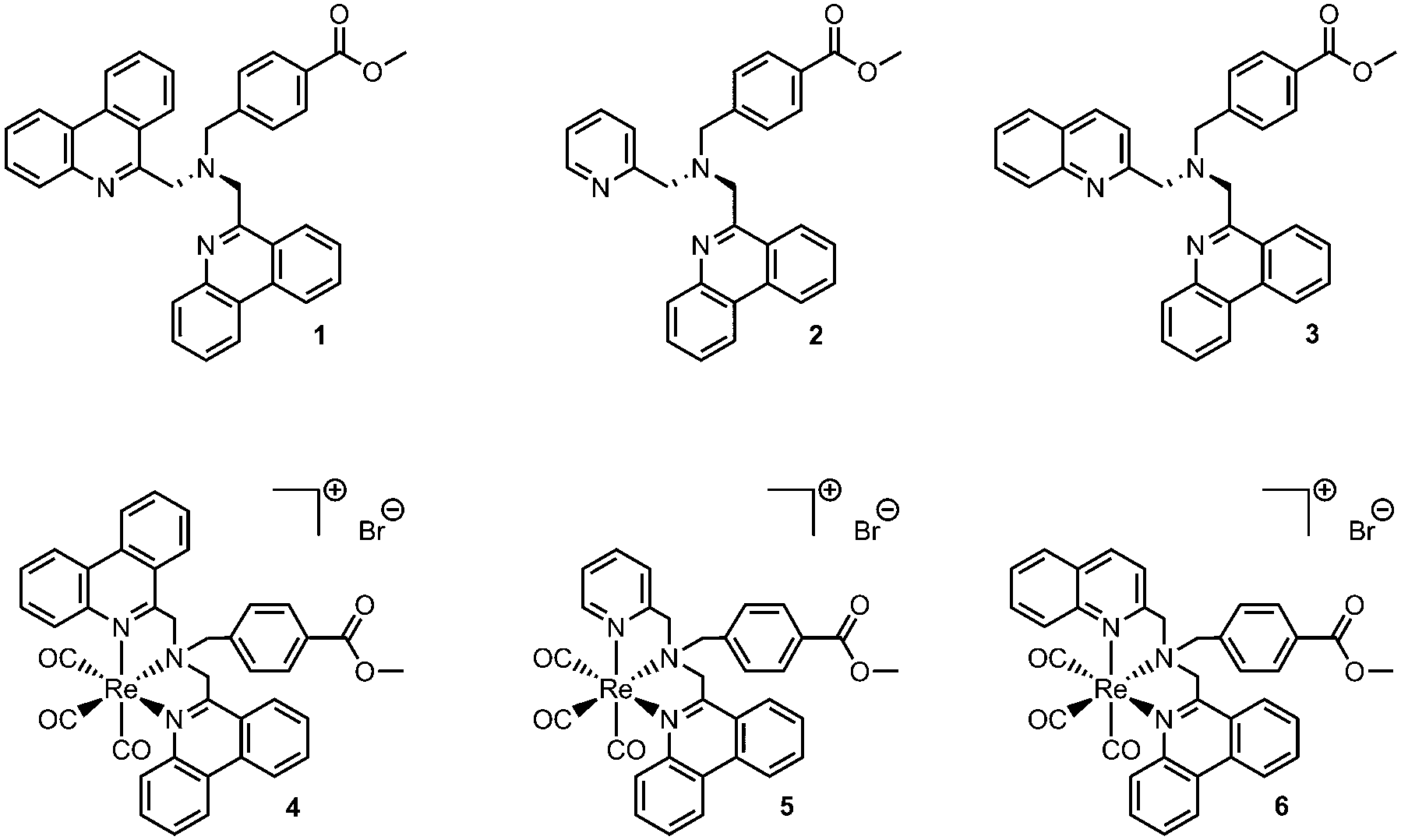 | ||
| Fig. 1 Novel tridentate ligands and their corresponding rhenium tricarbonyl complexes. | ||
All rhenium complexes show a characteristic broad UV/Vis absorption in the range between 300 nm and 380 nm. These absorption bands are predominantly defined by the phenanthridinyl moiety and are assigned to intraligand (IL) π → π* transitions (Fig. 2).29–31 The energy of those ligand-centered absorption bands do not vary considerably compared to pure ligands, but a substantial 3–4 fold enhancement of ε values is observed upon complexation of the fac-Re(CO)3 core (Fig. 2). The less intense and lower energetic MLCT band in rhenium complexes is typically localized adjacent to the IL transition, usually in the range of ∼350–500 nm.29,32,33 To further investigate the character of the transitions involved during excitation, we performed TDDFT calculations at the PBE0/6-31G(d)/LANL2DZ level of theory. Our results indicate that a description of the broad absorption bands as mixed MLCT/IL/ILCT represents a more precise description (see the ESI† for computational details and electron density difference maps).
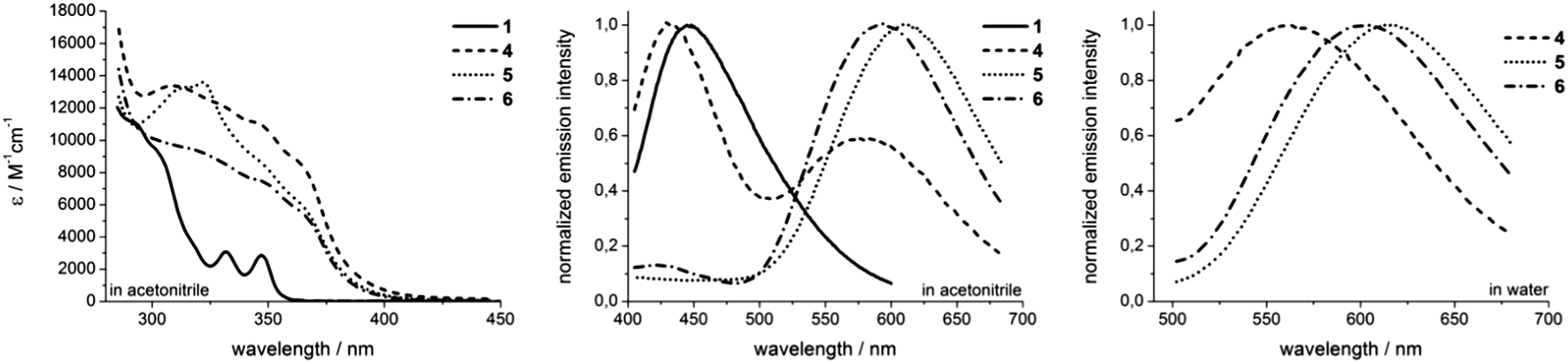 | ||
| Fig. 2 Absorption spectra of ligand 1 and rhenium complexes 4–6 in acetonitrile (left). Normalized emission spectra in acetonitrile (middle) and in water (right). All spectra were recorded at room temperature in not degassed solvents. | ||
Upon excitation at 350 nm, the complexes commonly emit in the orange range (λmax ≥ 560 nm) of the visible spectrum, giving large Stokes shifts of 210–250 nm (Fig. 2). These broad and structureless emission bands are most likely attributed to 3MLCT phosphorescence, frequently observed for rhenium tricarbonyl complexes.33 The solvent-dependent emission maxima of novel rhenium complexes shift in the order of λmax,4 < λmax,6 < λmax,5. Solvatochromic effects are contrary between the symmetrical complex 4 and the asymmetrical compounds 5 and 6. Whereas complex 4 exhibits a hypsochromic shift (λmax = 580 nm → λmax = 564 nm) upon changing from acetonitrile to water, both asymmetric complexes emit at longer wavelengths, performing bathochromic shifts of 9 nm upon switching from acetonitrile to aqueous solutions (Fig. 2 and Table 1). The latter may be explained by an increase of the dipole moment upon excitation, which consequently leads to a better stabilization of formed excited triplet states in more polar solvents,34 which however is inverse in the case of 4. Another significant alteration in emission behaviour between 4 and the asymmetric complexes 5 and 6 is that 4 displays two distinct emission bands upon excitation at 350 nm (Fig. 2). Due to the similarity of the higher energy emission of 4 to the emission observed for ligand 1 (Fig. 2), it can be assumed that this particular luminescence band stems from excited ligand-centered states.
| Compound | Solvent | Emission | Absorption | ||
|---|---|---|---|---|---|
| λ max [nm] | Φ [%] | τ [μs] | λ max [nm]/ε [L mol−1 cm−1] | ||
| a At 298 K. b Mean value and standard deviation from triplicates. c Estimated errors ± 15%. Contributions of amplitudes in bi-exponential fits are % values in brackets. | |||||
| 4 | MeCN | 580 | 0.12 ± 0.03 | 0.46 (2.4%), 0.09 (97.6%) | 310 (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400); 350 (10600) 400); 350 (10600) |
| MeCN (degassed) | 580 | 1.63 ± 0.19 | 1.24 (1.5%), 0.10 (98.5%) | ||
| H2O | 564 | 0.59 ± 0.20 | 0.42 (37.2%), 0.05 (62.8%) | ||
| 5 | MeCN | 609 | 0.12 ± 0.01 | 0.45 | 310 (9700); 350 (7300) |
| MeCN (degassed) | 609 | 0.29 ± 0.03 | 1.87 | ||
| H2O | 618 | 0.149 ± 0.001 | 0.61 | ||
| 6 | MeCN | 593 | 0.18 ± 0.01 | 0.58 | 310 (13100), 350 (8200) |
| MeCN (degassed) | 593 | 0.43 ± 0.06 | 3.01 | ||
| H2O | 602 | 0.209 ± 0.002 | 1.08 | ||
Quantitative characterizations of the photophysical properties of complexes 4–6 show firstly typical quantum yields in non-degassed acetonitrile in the range of 0.1–0.2%.23,25,35–37 Simultaneously, an increase of quantum yields is observed upon switching from aerated to degassed acetonitrile. The most obvious enhancement of Φ (almost 14-fold) upon switching to degassed acetonitrile is observed for complex 4, whereas just 2–3 fold increased quantum yields are found for the asymmetric complexes 5 and 6. When switching to aqueous solutions, the quantum yields increase by around 20% in the case of asymmetric complexes and a 5-fold enhanced Φ value of 0.59% is found for complex 4 (Table 1). Especially for complex 4 this could be attributed to the observed 3IL emission character. The influence of such a state seems to be much less pronounced in 5 and 6. Secondly, the decay in phosphorescence could be fitted to monoexponential decay curves for both asymmetric complexes 5 and 6. In contrast, the application of biexponential decay functions for correct adaptions of phosphorescence decays was necessary in the case of 4. From these curves, the emission lifetimes have been calculated (Table 1). Complex 6 displays the longest phosphorescence lifetimes, which are dependent on the solvent as well as on the presence or absence of dioxygen. In aerated acetonitrile, the 3MLCT emission lifetime is 580 ns, which increases to 3 μs in the absence of dioxygen. For complex 5, values of 450 ns and 1.87 μs have been obtained for aerated and degassed acetonitrile, respectively. Both asymmetric complexes are characterized by prolonged phosphorescence lifetimes in aqueous solutions with values of 1.08 μs for 6 and 609 ns for 5. Since the application of multiexponential decay functions is an indication for the involvement of more than just one emissive state,34 it can be assumed that complex 4 is not a pure 3MLCT emitter. Instead, it is likely that additional excited triplet states, e.g.3IL, influence the emission behaviour. The obtained values reveal that short lifetime emission (∼90–100 ns) is predominant in acetonitrile solutions, whereas long-lived contributions of 460 ns (aerated acetonitrile) and 1.24 μs (degassed acetonitrile) are mostly negligible. By switching to aqueous solutions the amount of short-lived emission (50 ns) decreases significantly, whereas the long-lived luminescence (420 ns) in turn increases.34 Overall, the luminescence lifetimes render these complexes interesting candidates for future advanced investigations in time-gated fluorescence microscopy or even fluorescence lifetime imaging (FLIM). Besides large Stokes shifts and long luminescence lifetimes, the evidence for emission from triplet states in rhenium complexes 4–6 is further underlined by the comparison of photophysical data obtained for aerated and degassed acetonitrile. Both quantum yields as well as phosphorescence lifetimes are decreased in the case of the aerated solvent, which is readily explained through quenching processes of the emissive triplet states by energy transfer to 3O2.32,38
With respect to the present photophysical data, ISC processes populating 3MLCT states seem to be more efficient in the case of asymmetric complexes. Whereas the excitation of the symmetric complex 4 populates fluorescence singlet states as well as mixed, radiative triplet states, 5 and 6 appear to be mostly 3MLCT emitters. The described rhenium complexes are characterized by solvatochromic effects, yielding improved luminescence characteristics in aqueous over acetonitrile solution. Although the solvent dependent emission maxima are contrary between 4 and asymmetric complexes, all rhenium compounds display increased quantum yields and at least the asymmetric compounds 5 and 6 demonstrate significantly prolonged emission lifetimes in aqueous solutions. The solvent dependency of emission behaviour of rhenium(I) tricarbonyl complexes has been shown to be largely influenced by the ligand structure. Although in most cases improved photophysical properties are obtained in less polar solvents,33 a few examples of opposite behaviour have been reported.15,39
With these promising photophysical data in hand, we went on to investigate the applicability of the rhenium complexes as imaging agents in live cell fluorescence microscopy. To this end, complexes 4–6 were added to IMIM-PC2 cancer cells in a concentration of 500 nM in supplemented DMEM and after incubation at 37 °C and 10% CO2 for 90 min, the cells were analysed by fluorescence microscopy. The large Stokes shift enabled the application of adjusted filter sets, which in turn resulted in images of high contrast. Good staining results were obtained for all three complexes upon excitation in the ultraviolet (λEx = 326–375 nm). As a note of caution, UV excitation is generally less preferred as it is potentially damaging for cells. Luminescence which is assigned to the 3MLCT emission (λEm > 510 nm) was observed, with accumulation in cytosolic, subcellular organelles (Fig. 3).
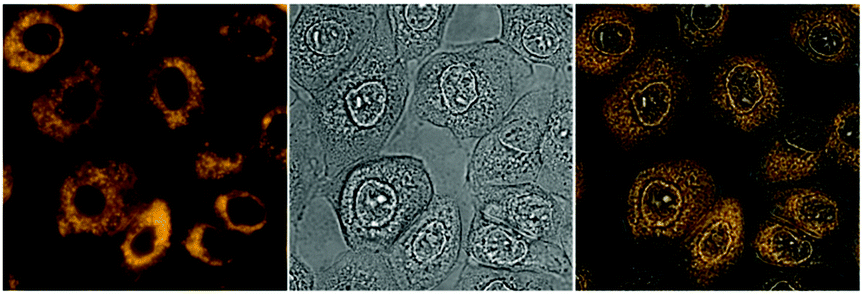 | ||
| Fig. 3 Imaging experiments with 6 (500 nm, 90 min incubation at 37 °C, 10% CO2) on IMIM-PC2 cells, left: fluorescence image (artificial colour), middle: bright field image, and right: merged image. | ||
To our great delight, we observed an accumulation of the complexes around the nucleus. Colocalisation studies (complexes 4 and 6) with commercially available organelle stains confirm the enrichment within the endoplasmic reticulum (ER). In contrast to the high degree of colocalization with the ER stain (Fig. 4), colocalisation experiments with Golgi and mitochondrially localizing dyes showed significant discrepancies, thus ruling out major colocalisation in either compartment. Since at least some selectivity of complexes for the ER has been demonstrated for IMIM-PC2 cells, we also carried out ER colocalization experiments on PT45, MCF-7, HT29, and Hep G2 cell lines. The results indicate a comparably high degree of matching regions. This confirms that selectivity for the ER is not specific for a single cell line.
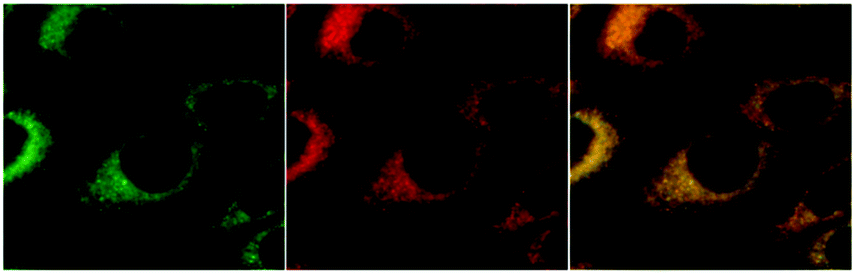 | ||
| Fig. 4 Colocalisation experiments with 6 on IMIM-PC2 cells, 600× magnification. Fluorescence images are coloured artificially. Left: 6, middle: commercial ER stain ER-ID™ red, and right: merged image. | ||
Further, unlike all commercially available organelle stains, all rhenium complexes displayed no indications of photobleaching throughout the experiments (see the ESI† for a comparison with commercial stains). Moreover, it can be stated that the asymmetric rhenium complex 6 demonstrated the most beneficial staining and luminescence properties with shortest exposure times and brightest images.
In summary, we have shown the successful synthesis of novel phenanthridinyl based chelating ligands and their rhenium(I) tricarbonyl complexes. Moreover, we have quantified their favourable photophysical properties which show advantages over previously published compounds with respect to the emission behaviour in aqueous media. The consistent photophysical behaviour in aqueous solutions combined with the fast cellular uptake makes the complexes studied herein not only suitable as imaging agents for conventional fluorescence microscopy, but also interesting candidates for future studies employing more sophisticated lifetime-based imaging techniques. Remarkably, because of the selective accumulation in the ER, these complexes are suitable for direct ER staining.
Notes and references
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 RSC.
- F. Wang, W. B. Tan, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, R1–R13 CrossRef CAS.
- K. K.-W. Lo, Struct. Bonding, 2007, 123, 205–245 CrossRef CAS.
- K. K.-W. Lo, K. Y. Zhang and S. P.-Y. Li, Eur. J. Inorg. Chem., 2011, 3551–3568 CrossRef CAS.
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed.
- J.-C. G. Buenzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef CAS PubMed.
- F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- M. V. Werrett, P. J. Wright, P. V. Simpson, P. Raiteri, B. W. Skelton, S. Stagni, A. G. Buckley, P. J. Rigby and M. Massi, Dalton Trans., 2015, 44, 20636–20647 RSC.
- M. V. Werrett, G. S. Huff, S. Muzzioli, V. Fiorini, S. Zacchini, B. W. Skelton, A. Maggiore, J. M. Malicka, M. Cocchi, K. C. Gordon, S. Stagni and M. Massi, Dalton Trans., 2015, 44, 8379–8393 RSC.
- C. A. Bader, R. D. Brooks, Y. S. Ng, A. Sorvina, M. V. Werrett, P. J. Wright, A. G. Anwer, D. A. Brooks, S. Stagni, S. Muzzioli, M. Silberstein, B. W. Skelton, E. M. Goldys, S. E. Plush, T. Shandala and M. Massi, RSC Adv., 2014, 4, 16345–16351 RSC.
- A. J. Amoroso, R. J. Arthur, M. P. Coogan, J. B. Court, V. Fernandez-Moreira, A. J. Hayes, D. Lloyd, C. Millet and S. J. A. Pope, New J. Chem., 2008, 32, 1097–1102 RSC.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernandez-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066–3068 RSC.
- M. P. Coogan, V. Fernandez-Moreira, J. B. Hess, S. J. A. Pope and C. Williams, New J. Chem., 2009, 33, 1094–1099 RSC.
- K. K.-W. Lo, M.-W. Louie, K.-S. Sze and J. S.-Y. Lau, Inorg. Chem., 2008, 47, 602–611 CrossRef CAS PubMed.
- Y. Shen, B. P. Maliwal and J. R. Lakowicz, J. Fluoresc., 2001, 11, 315–318 CrossRef CAS.
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed.
- C.-C. Ko, C.-O. Ng and S.-M. Yiu, Organometallics, 2012, 31, 7074–7084 CrossRef CAS.
- A. F. Armstrong, N. Oakley, S. Parker, P. W. Causey, J. Lemon, A. Capretta, C. Zimmerman, J. Joyal, F. Appoh, J. Zubieta, J. W. Babich, G. Singh and J. F. Valliant, Chem. Commun., 2008, 5532–5534 RSC.
- M. Bartholoma, J. Valliant, K. P. Maresca, J. Babich and J. Zubieta, Chem. Commun., 2009, 493–512 RSC.
- K. P. Maresca, S. M. Hillier, F. J. Femia, C. N. Zimmerman, M. K. Levadala, S. R. Banerjee, J. Hicks, C. Sundararajan, J. Valliant, J. Zubieta, W. C. Eckelman, J. L. Joyal and J. W. Babich, Bioconjugate Chem., 2009, 20, 1625–1633 CrossRef CAS PubMed.
- K. A. Stephenson, S. R. Banerjee, T. Besanger, O. O. Sogbein, M. K. Levadala, N. McFarlane, J. A. Lemon, D. R. Boreham, K. P. Maresca, J. D. Brennan, J. W. Babich, J. Zubieta and J. F. Valliant, J. Am. Chem. Soc., 2004, 126, 8598–8599 CrossRef CAS PubMed.
- N. Viola-Villegas, A. E. Rabideau, M. Bartholomä, J. Zubieta and R. P. Doyle, J. Med. Chem., 2009, 52, 5253–5261 CrossRef CAS PubMed.
- L. Wei, J. W. Babich, W. Ouellette and J. Zubieta, Inorg. Chem., 2006, 45, 3057–3066 CrossRef CAS PubMed.
- I. Kitanovic, S. Can, H. Alborzinia, A. Kitanovic, V. Pierroz, A. Leonidova, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wölfl and G. Gasser, Chem. – Eur. J., 2014, 20, 2496–2507 CrossRef CAS PubMed.
- G. Gasser, A. Pinto, S. Neumann, A. M. Sosniak, M. Seitz, K. Merz, R. Heumann and N. Metzler-Nolte, Dalton Trans., 2012, 41, 2304–2313 RSC.
- L. Raszeja, A. Maghnouj, S. Hahn and N. Metzler-Nolte, ChemBioChem, 2011, 12, 371–376 CrossRef CAS PubMed.
- L. Sacksteder, A. P. Zipp, E. A. Brown, J. Streich, J. N. Demas and B. A. DeGraff, Inorg. Chem., 1990, 29, 4335–4340 CrossRef CAS.
- A. Juris, S. Campagna, I. Bidd, J. M. Lehn and R. Ziessel, Inorg. Chem., 1988, 27, 4007–4011 CrossRef CAS.
- M. Norek, J. Dresner and J. Prochorow, Acta Phys. Pol., A, 2003, 104, 425–439 CrossRef CAS.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- R. A. Kirgan, B. P. Sullivan and D. P. Rillema, Top. Curr. Chem., 2007, 281, 45–100 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- L. Wei, J. Babich, W. C. Eckelman and J. Zubieta, Inorg. Chem., 2005, 44, 2198–2209 CrossRef CAS PubMed.
- M.-W. Louie, H.-W. Liu, M. H.-C. Lam, T.-C. Lau and K. K.-W. Lo, Organometallics, 2009, 28, 4297–4307 CrossRef CAS.
- A. O. T. Patrocinio, M. K. Brennaman, T. J. Meyer and N. Y. Murakami Iha, J. Phys. Chem. A, 2010, 114, 12129–12137 CrossRef CAS PubMed.
- A. A. Abdel-Shafi, J. L. Bourdelande and S. S. Ali, Dalton Trans., 2007, 2510–2516 RSC.
- B. Manimaran, P. Thanasekaran, T. Rajendran, R.-J. Lin, I. J. Chang, G.-H. Lee, S.-M. Peng, S. Rajagopal and K.-L. Lu, Inorg. Chem., 2002, 41, 5323–5325 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Expt. details; synthesis and characterizing data of the new compounds; crystallographic data of 4, 5 and 6 (CCDC 853881-853883); cell culture and fluorescence microscopy experiments. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc07553c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |