
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Positional effects of fluorination in conjugated side chains on photovoltaic properties of donor–acceptor copolymers†
Jisoo
Shin‡
,
Min
Kim‡
,
Jaewon
Lee
,
Heung Gyu
Kim
,
Hyeongjin
Hwang
and
Kilwon
Cho
*
Department of Chemical Engineering, Pohang University of Science and Technology, Pohang, 790-784, Korea. E-mail: kwcho@postech.ac.kr
First published on 19th December 2016
Abstract
The position at which conjugated side chains were fluorinated, the meta- or ortho-position in phenyl side chains, was varied to investigate the positional effects of fluorination on the energy levels, crystalline ordering, and photovoltaic properties of the polymers. The fluorine in the ortho-position achieved a lower HOMO energy level than that in the meta-position, but reduced the chain rigidity.
Conjugated polymers designed using the donor–acceptor (D–A) approach have tunable optical absorption bands, tunable molecular energy levels, and tunable carrier mobilities. For these reasons, conjugated polymers have been used extensively to develop organic solar cells (OSCs) with high power conversion efficiencies (PCEs).1,2 Control over the molecular energy levels, including the highest occupied molecular orbital (HOMO) level and the lowest unoccupied molecular orbital (LUMO) level, presents one of the most important challenges in the design of D–A polymers. Such control tunes the physical processes during exciton generation and diffusion, charge separation, and transport.3
Two strategies have been used to tune the molecular energy levels. One approach involves copolymerizing D–A polymers with different donor and acceptor building blocks,4,5 and the other involves introducing electron-withdrawing substituents onto the acceptor moieties to reduce the HOMO and LUMO levels without significantly altering the optical band gap.6,7 The introduction of conjugated side chains appears to be an effective means of controlling energy levels and intensifying the absorption properties and charge transport in D–A conjugated polymers, thereby improving the device performance in OSCs.8,9 Recently, our group reported that phenyl (Ph) side chains reduced the HOMO level and increased the open circuit voltage (Voc) more than did the thienyl side chain. Furthermore, symmetric phenyl side chains facilitated intramolecular and intermolecular charge carrier transport, thereby yielding a high short-circuit current (Jsc) by improving the intermolecular packing.10
Fluorination is a powerful approach to manipulate the energy level, molecular conformation, and eventually photovoltaic properties in organic solar cells. Although the relationship between the fluorine substitution in conjugated polymer backbones and molecular characteristics have been studied intensively, the effect of fluorine substitution onto conjugated side chains has not been studied yet. In this study, we examined the effects of fluorination in a conjugated side chain at various positions by synthesizing a new donor moiety with a fluorine atom substituted in place of a hydrogen atom at the ortho-position of alkoxy-phenyl (Ph-o-F) and evaluated with the fluorine substituted for hydrogen at the meta-position in alkoxy-phenyl (Ph-m-F). Benzodithiophene (BDT) derivatives with fluorine in the meta- or ortho-positions of the phenyl side chain were copolymerized with difluoro-benzothiadiazole (2FBT) (Scheme 1). Both copolymers, PPh-m-F and PPh-o-F, yielded high Voc values by reducing the HOMO energy level. In particular, introducing fluorine into the phenyl side chain near the backbone achieved a lower HOMO energy level when the fluorine was in the ortho-position than when it was in the meta-position, whereas the substitution of fluorine at the ortho-position reduced the chain rigidity and caused inferior crystalline ordering. These results suggested a mechanism underlying the effectiveness of fluorination at each position in the side chains. The Stille coupling reaction was used to synthesize the two types of polymers with fluorinated phenyl side chains on the BDT–2FBT backbone. The conditions and procedures used to synthesize the monomers and polymers are provided in detail in the ESI† (Schemes S1 and S2). PPh-m-F was characterized by a number-average molecular weight ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 12
n = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 Da and a polydispersity index (PDI) = 2.33; PPh-o-F was characterized by n = 15800 and PDI = 1.50 (Table 1).
300 Da and a polydispersity index (PDI) = 2.33; PPh-o-F was characterized by n = 15800 and PDI = 1.50 (Table 1).
 | ||
| Scheme 1 Molecular designs with different positions of fluorination. PPh* structure is referred to ref. 10. | ||
| Polymers |
n
(kDa)/PDI |
λ soln.max [nm] | λ filmmax [nm] | HOMO/LUMOb [eV] | E calg[eV] | E optg [eV] | T d [°C] |
|---|---|---|---|---|---|---|---|
| a Determined by GPC using polystyrene standards and 1,2-dichlorobenzene (o-DCB) as an eluent. b Measured from CV. c Onset of the UV-Vis spectra measured from thin films. d Measured from TGA (Fig. S2, ESI). Result of PPh* is referred to ref. 10 | |||||||
| PPh* | 14.1/1.17 | 577 | 654 | −5.23/−3.51 | 1.88 | 1.72 | 406 |
| PPh-m-F | 12.3/2.33 | 585 | 597 | −5.56/−3.79 | 1.92 | 1.77 | 422 |
| PPh-o-F | 15.8/1.50 | 543 | 588 | −5.70/−3.87 | 1.91 | 1.83 | 424 |
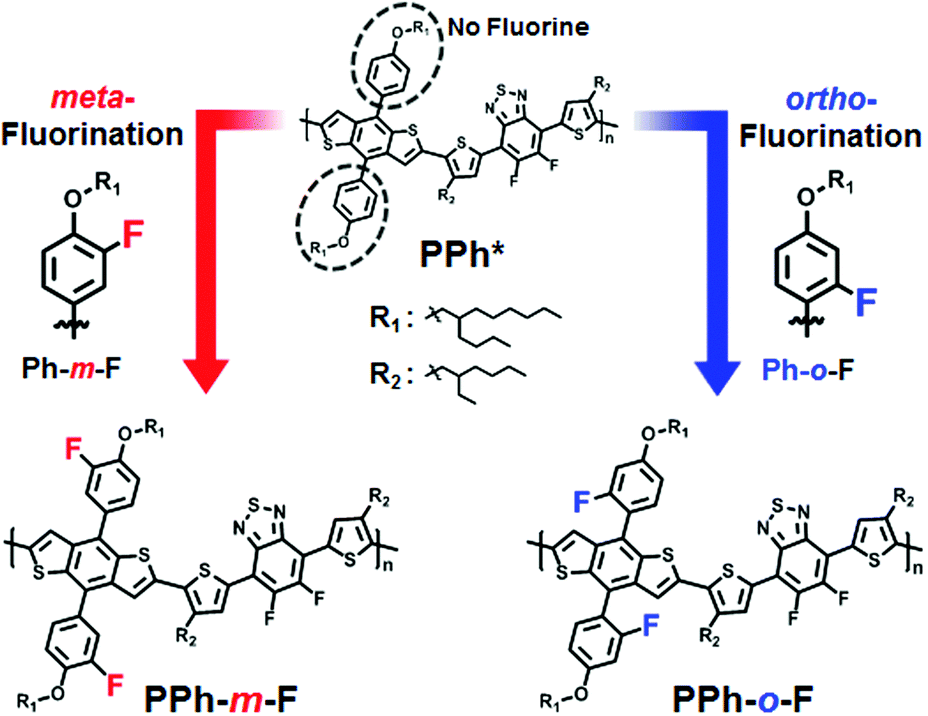
The HOMO energy levels were found to be −5.56 eV in PPh-m-F and −5.70 eV in PPh-o-F using cyclic voltammetry (CV) (Fig. 1a). Compared to our previous studies using a non-fluorinated phenyl side chain (PPh),10 the introduction of a fluorine atom into the phenyl side chain decreased the HOMO energy level to an extent comparable to that obtained by substituting electron-withdrawing groups into the polymer chain backbone.6,7 This result suggested that the presence of strongly electron-withdrawing fluorine atoms in the side chain of the polymer reduced the HOMO energy level. Furthermore, fluorination at the ortho-position of the phenyl side chain reduced the HOMO energy level to a greater extent than did fluorination at the meta-position. The resonance structures predicted that the ortho-fluoro-phenyl substituted BDT should have a lower HOMO energy level than the meta-fluoro-phenyl substituted BDT (Fig. S2, ESI†). The substitution of the fluorine atom into the meta-position yielded an unstable carbanion with lone pair electrons on fluorine. In this case, the HOMO energy level of the meta-fluoro-phenyl BDT was elevated. The fluorination of the ortho-position reduced the HOMO level because the carbanion was stabilized by delocalization.
 | ||
| Fig. 1 Cyclic voltammograms of the PPhs in the CH3CN solutions at a scan rate of 50 mV s−1. Result of PPh* is referred to ref. 10. (b) Normalized UV-Vis absorption spectra of the polymers in DCB at a concentration of 0.025 g L−1 and thin films. | ||
The UV-Vis absorption spectra (Fig. 1b) of both the polymers featured two absorption bands, one at short wavelengths (350 ≤ λ ≤ 450 nm), which originated from the π–π* transitions, and a broad band at 550 ≤ λ ≤ 650 nm due to intramolecular charge transfer. Similar results have been reported for other D–A conjugated copolymers.11,12 The optical band gaps of PPh-m-F and PPh-o-F were 1.77 eV and 1.83 eV, respectively (Table 1). Variations in the band edges were attributed to the backbone planarity and intermolecular packing. The electron affinities of the substituents in the side chains were related to the band edges.13,14 The solution spectra of PPh-m-F revealed a pronounced shoulder at long wavelengths as a result of strong aggregation related to polymer interactions, possibly due to the planar backbone conformations, which increased the intermolecular interaction strength.15 PPh-o-F showed good solubility and no shoulder peaks in the solution state. In the solid film, PPh-m-F showed a strong vibronic structure adjacent to the absorption maximum. The interchain π–π* transitions were strong, indicating that this polymer formed structurally organized and orderly packed structures in the thin film state. Likewise, PPh-o-F had a lower absorption coefficient than did PPh-m-F over the entire UV-Vis range (Fig. S3, ESI†). Consequently, the broadened and extended absorption spectra of PPh-m-F resulted in enhanced Jsc and PCE.
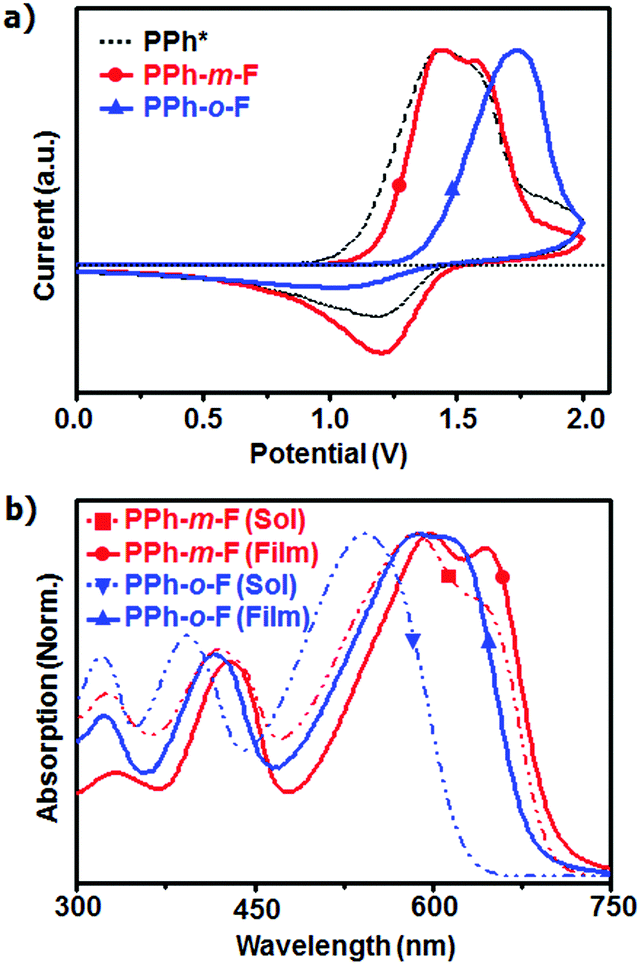
To obtain insight into how the position of the fluorine atom in the conjugated side chains affected the molecular conformations and electronic properties of the polymers, density functional theory (DFT) calculations were performed via Gaussian simulations using the B3LYP/6-31G model.16,17 The calculations were performed using optimized moieties and three repeating units to predict the superposition of the HOMOs and the LUMOs (Fig. S4, ESI†). The HOMO orbitals were delocalized along the chain backbone. The fluorine atom was closer to the HOMO orbitals in the PPh-o-F backbone than in the PPh-m-F backbone (Fig. S5a, ESI†). The fluorine atom in PPh-m-F was one σ bond further from the backbone than the fluorine atom in PPh-o-F (Fig. S5b, ESI†). The inductive effect depends critically on the distance between groups through σ bonds, so PPh-o-F may display a lower HOMO level in the CV results than PPh-m-F.
The dihedral angle θa in the backbone bridge joint between the BDT unit and the 2FBT unit in the first repeating unit was 21.5° in the PPh-m-F model but 25.2° in PPh-o-F (Fig. 2b and Table S1, ESI†). Compared to PPh-m-F, the backbone of PPh-o-F was less planar and the structure along the π–π direction was less bulky, with a backbone plane that was more extensively rotated. The results obtained from the dihedral angles agreed well with the lack of shoulder peaks in the UV-Vis absorption and the lower absorption coefficient of PPh-o-F compared to that of PPh-m-F. The backbone of PPh-o-F may be easily twisted, and the resulting restricted intermolecular chain packing may improve the solubility. The dihedral angles θb calculated between the plane of the phenyl side chain and the plane of the BDT was 56.7° in PPh-m-F and 62.1° in PPh-o-F. The large plane-to-plane torsional angle in PPh-o-F arose from the steric hindrance between the fluorine atom on the phenyl group and the H atom in the neighboring BDT. An effective overlap between the orbitals perpendicular to the backbone was, therefore, impossible (Fig. 2c). The substitution of the fluorine atoms for hydrogen atoms in the conjugated side chains of the polymers significantly influenced the crystal and electrical properties of the polymers.
 | ||
| Fig. 2 Illustrations for the chain conformations using the DFT calculation (B3LYP/6-31G) model. The yellow, red, blue and sky blue symbols represent sulfur, oxygen, nitrogen, and fluorine atoms, respectively. (a) Top view of polymer chain. (b) Side view of PPh-m-F and PPh-o-F. θa is a key dihedral angle determining the backbone planarity. (c) Conjugated region in chain direction and side chain direction. | ||
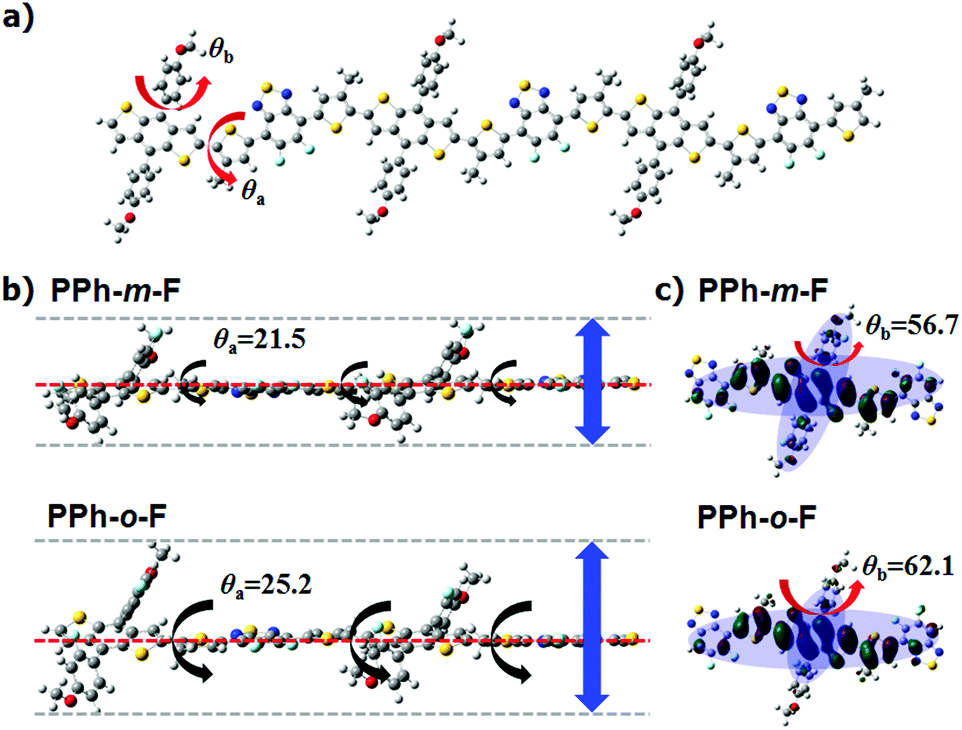
The effects of the fluorination position on the crystal structure and molecular orientation were measured using two-dimensional grazing-incidence X-ray diffraction (2D GIXD) analysis and atomic force microscopy (AFM).18 The substitution of the fluorine atoms for hydrogen at the meta-position of the phenyl side chain yielded a more distinct crystalline feature in the pristine polymer film, with high-order (h00) and strong diffraction peaks and edge-on crystallites oriented predominantly along the out-of-plane direction (Fig. 3). The π–π stacking distance was shorter in PPh-m-F (0.38 nm) than in PPh-o-F (0.46 nm). This difference indicated that the chain of PPh-m-F was more planar and was packed closely along the π–π direction. The GIWAXS results obtained from PPh-m-F and PPh-o-F were correlated with the crystalline properties observed in the UV-Vis spectra and were supported by the DFT calculations. The AFM images revealed that the morphologies were distinctly affected by the position of the fluorination. The PPh-o-F film was smooth and flat, whereas the PPh-m-F film was rough and had large interconnected grains. All of these results suggested that PPh-m-F included reinforced intermolecular interactions that increased the number of interconnected crystalline grains and, as a result, presumably increased the charge-transport properties compared to those of PPh-o-F.
 | ||
| Fig. 3 (a) GIWAXS data of homogeneous polymer films of PPh-m-F and PPh-o-F. The insets are AFM height images of neat polymer films of PPh-m-F and PPh-o-F prepared by DCB. (b) The corresponding 1D GIWAXS diffractogram profiles along the out-of-plane direction and in-plane direction. | ||
The photovoltaic characteristics of both the polymers were evaluated by fabricating conventional cells (ITO/MoO3/polymer:PC71BM/LiF/Al) using the synthesized polymers. The device performances were optimized by using 1,8-diiodooctane (DIO) as a solvent additive and by fabricating the devices in various thicknesses and weight ratios of polymers and PC71BM.19,20 The photovoltaic efficiencies differed significantly, depending on the position of the F atom in the side chain. The device that used PPh-m-F yielded Jsc = 11.25 mA cm−2, FF = 64.6%, Voc = 0.91 V, and PCE = 6.62%. An otherwise-identical device that used PPh-o-F displayed inferior electrical characteristics (Jsc = 4.31 mA cm−2, FF = 60.6%, Voc = 0.96 V, and PCE = 2.51%) under the same conditions (Fig. 4a, Table 2 and Table S2, ESI†). The Voc of PPh-o-F device was higher than that of PPh-m-F showing similar tendency as the CV results. However, the PPh-o-F device showed higher bimolecular recombination than that of the PPh-m-F device due to their differences in morphological and electrical properties, which could reduce achievable photovoltage of the PPh-o-F device (Fig. S6, ESI†). Over the range of 300 ≤ λ ≤ 800 nm, the device prepared with PPh-m-F achieved a maximum EQE > 70% (at λ = 400 nm) and, therefore, displayed a higher Jsc than did the device prepared with PPh-o-F (Fig. 4b). The device based on PPh-m-F displayed higher values of Jsc and FF than did the PPh-o-F based device. The difference was attributed to differences between the light absorption abilities, the charge carrier mobilities and the morphological structures of the active layers.
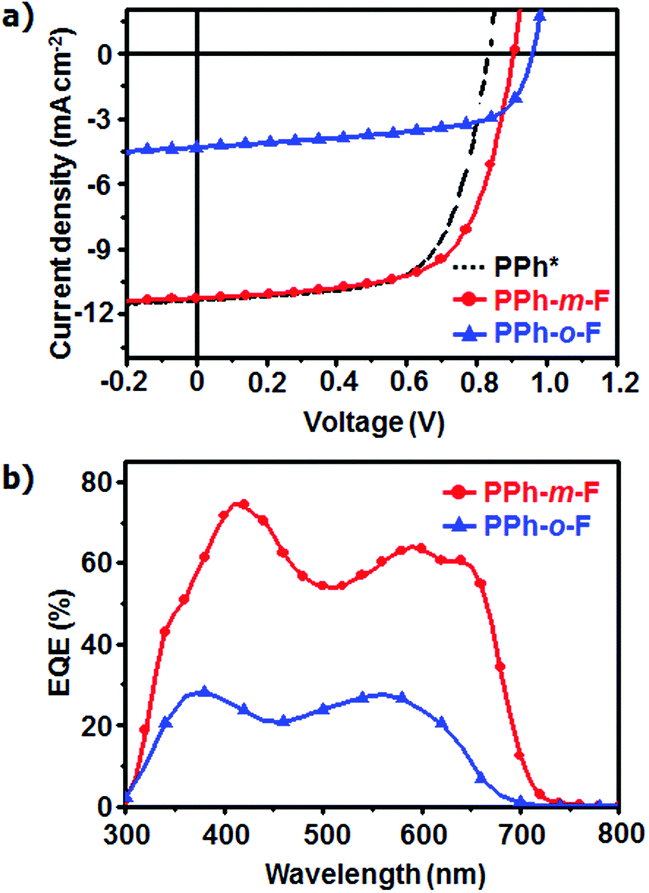 | ||
| Fig. 4 (a) J–V characteristics of PPhs/PC71BM solar cell prepared by DCB with DIO under illumination of AM 1.5 G, 100 mW cm−2. Result of PPh* is referred to ref. 10. (b) EQE curves of the corresponding organic solar cells. | ||
:PC71BM were fixed at 1:1.2 (w:w) with DIO 1.5 v% in DCB. Result of PPh* is referred to ref. 10. Performance metrics are average numbers from eight devices of each type under the illumination conditions of AM 1.5 G, 100 mW cm−2. SCLC results measured by hole only devices. Standard deviation is included in the ESI
| Polymer | V oc [V] | J sc [mA cm−2] | FF [%] | PCEmax (avg) [%] | Hole mobility [cm2 V−1 s−1] |
|---|---|---|---|---|---|
| PPh* | 0.83 | 11.33 | 66.3 | 6.23 (6.02) | 2.50 × 10−4 |
| PPh-m-F | 0.91 | 11.25 | 64.6 | 6.62 (6.07) | 1.13 × 10−4 |
| PPh-o-F | 0.96 | 4.31 | 60.6 | 2.51 (2.43) | 5.64 × 10−5 |
A 2D-GIXD analysis of the PPh-m-F:PC71BM blend film detected prominent higher-order (h00) diffraction peaks that were attributed to effective intermolecular packing due to the high planarity of the polymers (Fig. S7, ESI†). The presence of high peaks may improve the space-charge-limited current (SCLC), indicating that the hole mobility in PPh-m-F was higher than that in PPh-o-F (Table 2). A high crystallinity increases charge transport in photovoltaic devices. The UV-Vis results and DFT calculations discussed above suggested that PPh-m-F included a planar backbone chain, which yielded fast charge transport in the polymer chain. Transmission electron microscopy (TEM) images showed that the PPh-m-F:PC71BM blend film had the relatively small domain size to induce efficient exciton dissociation at the polymer:PC71BM interface, hence increasing Jsc (Fig. S8, ESI†). Therefore, the PPh-m-F:PC71BM blend film had advantageous characteristics for photovoltaic performances compared to the PPh-o-F:PC71BM blend film.
The introduction of a fluorine atom into the conjugated side chain of the polymer increased Voc by effectively lowering the HOMO energy level. PPh-m-F and PPh-o-F showed quite different optoelectronic characteristics as a result of the position of fluorine in the conjugated side chains. A fluorine atom positioned at the ortho- of the phenyl side chain lowered the HOMO more powerfully and induced better miscibility with PC71BM than did the fluorine at the meta-position, although the crystalline properties were degraded. Well-developed polymer crystals were formed only when the polymer included a fluorine atom at the meta-position of the phenyl side chain. Good chain planarity increased the light absorption and appeared to improve charge transport. As a result, the introduction of a fluorine atom at specific positions in the side chain provides a promising method for controlling both Jsc and Voc by modulating the intermolecular packing and molecular energy levels. This easy and effective strategy demonstrates the utility of side chain applications and may improve the photovoltaic efficiencies of optoelectronic devices.
This work was supported by a grant (Code No. 2011-0031628) from the Center for Advanced Soft Electronics under the Global Frontier Research Program of the Ministry of Science, ICT and the Future Planning, Korea. The authors thank the Pohang Accelerator Laboratory for providing the synchrotron radiation sources at the 3C, 9A beam lines used in this study.
Notes and references
- A. Ajayaghosh, Chem. Soc. Rev., 2003, 32, 181–191 RSC.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- X. F. Liu, W. Wen and G. C. Bazan, Adv. Mater., 2012, 24, 4505–4510 CrossRef CAS PubMed.
- I. Osaka, T. Kakara, N. Takemura, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 8834–8837 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- H.-H. Chang, C.-E. Tsai, Y.-Y. Lai, W.-W. Liang, S.-L. Hsu, C.-S. Hsu and Y.-J. Cheng, Macromolecules, 2013, 46, 7715–7726 CrossRef CAS.
- Y. Wang, F. Yang, Y. Liu, R. Peng, S. Chen and Z. Ge, Macromolecules, 2013, 46, 1368–1375 CrossRef CAS.
- R. S. Kularatne, F. J. Taenzler, H. D. Magurudeniya, J. Du, J. W. Murphy, E. E. Sheina, B. E. Gnade, M. C. Biewer and M. C. Stefan, J. Mater. Chem., 2013, 1, 15535–15543 RSC.
- J. Shin, M. Kim, J. Lee, D. Sin, H. G. Kim, H. Hwang and K. Cho, RSC Adv., 2015, 5, 106044 RSC.
- J. Lee, S. B. Jo, M. Kim, H. G. Kim, J. Shin, H. Kim and K. Cho, Adv. Mater., 2014, 26, 6706–6714 CrossRef CAS PubMed.
- T. L. Nguyen, H. Choi, S.-J. Ko, M. A. Uddin, B. Walker, S. Yum, J.-E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040–3051 CAS.
- L. Huo, L. Ye, Y. Wu, Z. Li, X. Guo, M. Zhang, S. Zhang and J. Hou, Macromolecules, 2012, 45, 6923–6929 CrossRef CAS.
- I. C. Wu, C. H. Lai, D. Y. Chen, C. W. Shih, C. Y. Wei, B. T. Ko, C. Ting and P. T. Chou, J. Mater. Chem., 2008, 18, 4297–4303 RSC.
- T. Q. Nguyen, I. B. Martini, J. Liu and B. J. Schwartz, J. Phys. Chem. B, 2000, 104, 237–255 CrossRef CAS.
- S. B. Darling and M. Sternberg, J. Phys. Chem. B, 2009, 113, 6215–6218 CrossRef CAS PubMed.
- Y.-X. Xu, C.-C. Chueh, H.-L. Yip, F.-Z. Ding, Y.-X. Li, C.-Z. Li, X. Li, W.-C. Chen and A. K.-Y. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS PubMed.
- W. Chen, M. P. Nikiforov and S. B. Darling, Energy Environ. Sci., 2012, 5, 8045–8074 CAS.
- H.-C. Liao, C.-C. Ho, C.-Y. Chang, M.-H. Jao, S. B. Darling and W.-F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385–391 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc06646a |
| ‡ Jisoo Shin and Min Kim contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |