
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green and highly efficient synthesis of perylene and naphthalene bisimides in nothing but water†
Bettina
Baumgartner
a,
Anastasiya
Svirkova
b,
Johannes
Bintinger
c,
Christian
Hametner
c,
Martina
Marchetti-Deschmann
b and
Miriam M.
Unterlass
*a
aTechnische Universität Wien, Institute of Materials Chemistry, Getreidemarkt 9/BC/2, A-1060 Vienna, Austria. E-mail: miriam.unterlass@tuwien.ac.at; Fax: +43-(0)1-58801-165981; Tel: +43-(0)1-58801-165206
bTechnische Universität Wien, Institute of Chemical Technologies and Analytics, Getreidemarkt 9/164-IAC, 1060 Vienna, Austria
cTechnische Universität Wien, Institute of Applied Synthetic Chemistry, Getreidemarkt 9/163, 1060 Vienna, Austria
First published on 25th November 2016
Abstract
High-purity, symmetrically substituted perylene and naphthalene bisimides were obtained by hydrothermal condensation of monoamines with the corresponding bisanhydride. The hydrothermal imidization proceeds quantitatively, without the need for organic solvents, catalysts or excess of the amines.
The development of chemical reactions that minimize environmental impact, as expressed by the principles of “green chemistry”, is currently one of the major challenges in synthetic chemistry.1 This effort is even more important for (i) widely encountered chemical functions, (ii) that are typically only accessible via harsh and harmful synthetic procedures. The imide aka diacyl amide function is among such omnipresent chemical moieties. Imides are widely used as protecting groups (e.g. phthalimides2), for bioconjugation reactions (e.g. maleimide–thiol coupling3–5), in polymer chemistry (e.g. polyimides,6,7 maleimide resins8), as dyes (e.g. perylene (PBIs) and naphthalene bisimides (NBIs)),9–11 and chromonic liquid crystals (perylene bisimide derivatives).12–14 The molecular features generating PBIs' and NBIs' dye properties, i.e. aromatic π-systems, also enable energy and electron transfer ability, and allow for the formation of intriguing supramolecular assemblies.10,11 Symmetrically N-substituted NBIs and PBIs are most commonly synthesized by (i) Langhals' method consisting of condensing the naphthalene (NBA) or perylene bisanhydride (PBA) with the respective monoamine in molten imidazole (or quinoline) employing zinc acetate or dicyclohexylcarbodiimide as promotor at high temperatures (180–230 °C).15–18 (ii) Moreover, the addition of Zn salts can be omitted using aprotic polar solvents (e.g. DMF) at high temperatures.11,19 Scarcely used alternative routes include the thermal solid-state condensation of co-crystals of NBA and amines at 160 °C,20 and direct approaches between bisanhydride and the liquid amine using acetic acid as promotor.21,22 While these techniques, especially Langhals' method, generate NBIs and PBIs in high yields, the products have to be purified chromatographically or by recrystallization. Moreover, typically 4-fold (in some cases up to 20-fold) molar excess of the monoamines are used, which is highly undesirable in terms of “green chemistry”. Clearly, for both NBIs and PBIs, these most common syntheses can be qualified as toxic and harsh. We recently reported that fully aromatic polyimides can be synthesized by so-called hydrothermal polymerization (HTP).23 Here, the imide functions are obtained by condensation of aromatic diamines with aromatic dianhydrides (or tetracarboxylic acids) in water at >180 °C and autogenous pressures. For instance, at 180–200 °C, the autogenous pressure of H2O is of only 12–17 bar. These rather moderate conditions can be generated easily in commercial autoclaves and do not require custom-made set-ups. In the hydrothermal regime, H2O becomes increasingly apolar and at the same time shows an increase in its ionic product, i.e. the concentration of both OH− and H3O+ ions increases, enabling the medium itself to act as both an acid and a base catalyst, which is indeed beneficial for organic condensations.24 These findings prompted us to investigate if HTP conditions could also be applied to small organic molecules.
In this contribution, we report a novel, green technique to prepare symmetrically substituted PBIs and NBIs in nothing but water. First, we reacted a series of eleven n-alkyl monoamines (n-Cn-NH2) with PBA (Fig. 1). Specifically, we employed n-C5-, n-C6-, n-C7-, n-C8-, n-C9-, n-C10-, n-C11-, n-C12-, n-C14-, n-C15, n-C16- and n-C18-NH2. Therefore, 1 eq. of PBA and 2 eq. of n-Cn-NH2 were stirred in deionized water (c = 0.03 mol L−1) at RT for 15 min. The resulting suspension was transferred to a non-stirred autoclave that was placed into an oven preheated at 200 °C. After 24 h, the autoclave was allowed to slowly cool back to RT, and the crude product was isolated by filtration, but not subjected to any further purification such as conventional recrystallization or chromatographic separation.
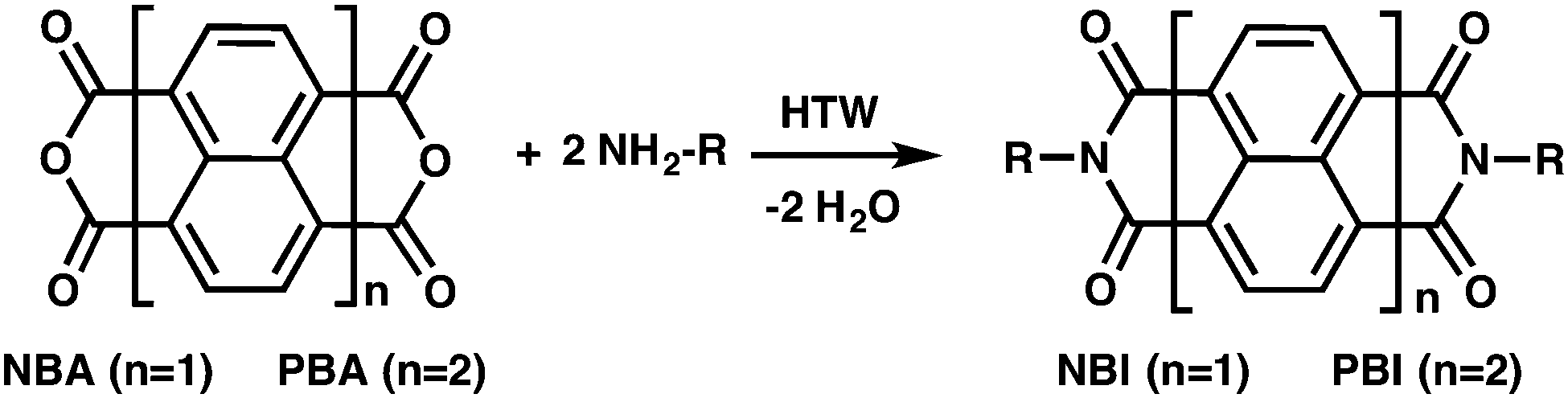 | ||
Fig. 1 Hydrothermal condensations of NBIs and PBIs: the dianhydrides NBA and PBA are reacted with different monoamines in equimolar ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, no excess of RNH2). HTW = high-temperature water, here: 200 °C and autogenous pressure. :2, no excess of RNH2). HTW = high-temperature water, here: 200 °C and autogenous pressure. | ||
Note that all n-Cn-PBI products, except n-C16- and n-C18-PBI, were obtained quantitatively. In the ATR-FTIR spectra of n-Cn-PBIs (ESI†), we find the for PBIs characteristic imide modes (1693 cm−1 and 1654 cm−1). Moreover, the NH2 modes of the n-alkylamines (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (N–H) and intermolecular H-bonding of amine groups ≈3330–3200 cm−1, 2–3 modes) are fully absent in all cases. n-C5-, n-C8-, n-C9-, n-C10-, n-C14- and n-C15-PBI show no remaining anhydride modes (as(C
(N–H) and intermolecular H-bonding of amine groups ≈3330–3200 cm−1, 2–3 modes) are fully absent in all cases. n-C5-, n-C8-, n-C9-, n-C10-, n-C14- and n-C15-PBI show no remaining anhydride modes (as(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) ≈ 1772 cm−1, s(CO) ≈ 1733 cm−1) related to unreacted PBA. Hence, ATR-FTIR analysis indicates both the full consumption of PBA and the absence of perylene monoimide monoanhydride, which would in principle be a possible product. Indeed, Nagao & Misono and Huang et al. have used H2O/ROH mixtures as solvents for the synthesis of perylene monoimide monoanhydrides, which were in both cases the major products (≈80%), and PBIs only formed in minority (≈20%).25,26 Given that these reports (and also Langhals' method) employ important excesses of the monoamines, it is most fascinating that fully condensed PBIs can be obtained hydrothermally without any additives at equimolar reactant ratio.
O) ≈ 1772 cm−1, s(CO) ≈ 1733 cm−1) related to unreacted PBA. Hence, ATR-FTIR analysis indicates both the full consumption of PBA and the absence of perylene monoimide monoanhydride, which would in principle be a possible product. Indeed, Nagao & Misono and Huang et al. have used H2O/ROH mixtures as solvents for the synthesis of perylene monoimide monoanhydrides, which were in both cases the major products (≈80%), and PBIs only formed in minority (≈20%).25,26 Given that these reports (and also Langhals' method) employ important excesses of the monoamines, it is most fascinating that fully condensed PBIs can be obtained hydrothermally without any additives at equimolar reactant ratio.
While the ATR-FTIR spectra of n-C6-, n-C7-, n-C11- and n-C12-PBIs show very small remaining CO anhydride modes, n-C16- and n-C18-PBI show considerable anhydride modes (see ESI†). The incompleteness of formation of the latter two PBIs is however not related to the basicity of n-C16 and n-C18-NH2, since the pKa values of n-alkylamines generally do not vary for different alkyl substituents, but all lie in the same range (10.4–10.6).27,28 The water-solubility is however strongly decreasing with increasing alkyl chainlength. Therefore, we suppose that the hydrothermal formations of n-C16- and n-C18-PBIs are less complete after 24 h, because of n-C16- and n-C18-NH2's limited solubility in H2O, and thus limited availability for reaction. The increase in apolarity of the n-alkylamines and hence also of the n-Cn-PBIs is indeed reflected by the aspect of the reaction mixtures after hydrothermal imidization: while the n-Cn-PBIs bearing short alkyl chains result as homogeneous dispersions, longer alkyl tails lead to a precipitate covered by a translucent aqueous phase. Specifically, n-C5 to n-C8-PBI are obtained as bright blood-red dispersions with no visible phase separation (photographs, see ESI†). n-C9 and n-C10-PBI form a ruby-red colored precipitate covered by a turbid red dispersion, and from n-C11-PBI onwards, the PBIs fully precipitate as brownish-red powders, covered by a translucent yellowish aqueous phase (ESI†).
Micromorphological investigations via scanning electron microscopy (SEM) confirm this trend: n-C5- and n-C6-PBI are cuboid-like particles of ca. 5–10 μm in length and 1–2 μm in thickness (see Fig. 2B and ESI†). In n-C7- and n-C8-PBI roundish aggregates of ca. 10–50 μm in diameter coexist with ribbons of ca. 10 μm × 100 nm × 1 μm (length × thickness × depth) (see Fig. 2C and ESI†). n-C9- to n-C15-PBI all form stacks of discoid platelets that are each ca. 5 μm in diameter and ca. 100 nm in thickness (see Fig. 2D and ESI†). For longer alkyl-chains in n-Cn-PBIs, the increased hydrophobicity generates the compounds' drive to limit the contact with the polar medium H2O: the hydrothermally formed PBIs minimize their surface area for a given volume by adopting roundish morphologies. In contrast, PBIs bearing short alkyl chains do not adopt such roundish morphologies: n-C5- and n-C6-PBI resemble the morphology of hydrothermally recrystallized PBA that we prepared for comparison (see Fig. 2A and ESI†). Overall, it can be stated that the PBI morphology is strongly influenced by the N-alkyl substitution.
 | ||
| Fig. 2 SEM images of hydrothermally synthesized PBIs and PBA. (A) Hydrothermally treated PBA (24 h, 200 °C). (B–D) n-Cn-PBIs obtained hydrothermally (24 h, 200 °C). B = n-C5-PBI, C = n-C8-PBI, D = n-C12-PBI. | ||
Despite the roundish morphologies found in n-C7 to n-C15, all PBIs are highly crystalline as evinced by powder X-ray diffraction (PXRD) measurements (ESI†). Since hydrothermal synthesis has been shown to yield outstanding crystallinity for polyimides,23,24,29 this result comes as no surprise. n-C16- and n-C18-PBI are also highly crystalline in PXRD, but their incompleteness of reaction (especially for n-C18-PBI) is further underlined by the presence of reflections corresponding to PBA (ESI†).
The N-substitution of PBIs with alkyl chains is a means to increase their solubility in organic solvents. Therefore, the n-Cn-PBIs could be analyzed by solution NMR for estimating their purity. Representative 1H-NMR spectra of n-C8- and n-C14-PBI are shown in Fig. 3 (see ESI† for larger representation and spectra of the other n-Cn-PBIs). Both spectra show all expected peaks and the found integral ratios correspond well to the theoretical ratios (n-C8-PBI, Ha:Hb:Hc:Hd:He expected: 6:20:4:4:8, found: 6.14:21.32:3.92:4.00:8.00; n-C14-PBI, Ha:Hb:Hc:Hd:He expected: 6:44:4:4:8, found: 6.21:47.16:3.87:4.11:8.00). While PBA is insoluble in the solvent used for NMR measurements (CDCl3/trifluoroacetic acid, 5/1 v/v), remaining monoamine would affect the integrals corresponding to CH2 and CH3 groups (Ha, Hb and Hc). It becomes clear from Fig. 3 that there is no unreacted monoamine present. Note that even for n-C16- and n-C18-PBI, we did not find residual amines in NMR (ESI†), despite the incompleteness, judged from ATR-FTIR, of the transformation in both cases. We believe, that the remaining amine was dissolved/dispersed in the aqueous supernatant after the HT transformation, most likely in its amminium form. Note that selected n-Cn-PBI were additionally confirmed by mass spectrometry (ESI†).
 | ||
| Fig. 3 1H-NMR spectra (600 MHz, CDCl3/TFA: 5/1 v/v, 16 scans) of n-Cn-PBIs synthesized hydrothermally at 200 °C for 24 h; black = n-C14-PBI, turquoise = n-C8-PBI. Solvent residual peak (7.26 ppm) is marked with *. | ||
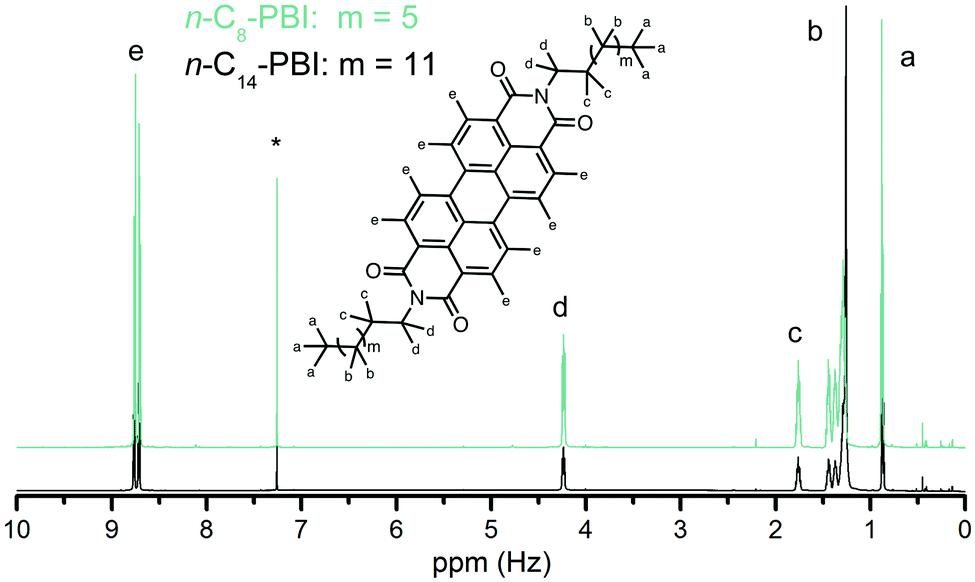
Soluble PBIs find tremendous interest for energy-related applications, e.g. in the field of organic electronics, and as fluorescence dyes.10 In both cases, their solubility is required for processing, and their purity is of prime importance. For testing the dye quality, we performed UV-Vis absorption and fluorescence measurements in CH2Cl2 and CHCl3. The UV-Vis and the fluorescence spectrum of n-C14-PBI are exemplarily shown in Fig. 4 (see ESI† for spectra of all other n-Cn-PBIs). Both the absorption maxima (526 nm, 490 nm, 458 nm) and the fluorescence maxima (533 nm, 574 nm, 624 nm) are in good agreement with the literature.9 The fluorescence quantum yields of all measured n-Cn-PBIs are ΦF ≈ 1 (see ESI†). All n-Cn-PBIs are soluble in a number of organic solvents (CH2Cl2,CHCl3, DMSO, DMF, benzene, nitrobenzene, in the mg/100 mL range) generating strongly colored, translucent solutions (Fig. 4, ESI†). Since the fluorescence can be affected by impurities that also show fluorescence, such as aromatics/heteroaromatics, the hydrothermal imide synthesis presented here is clearly preferable to condensations in imidazole, especially since it requires no purification.
 | ||
| Fig. 4 Left: UV-Vis (black) and fluorescence (turquoise) spectrum of n-C14-PBI in CH3Cl synthesized hydrothermally at 200 °C for 24 h. Right: Aspect of n-C14-PBI under visible (top) and UV-light (bottom) in the solvents (from left to right): CHCl3, CH2Cl2, DMF, DMSO, THF, TFA. | ||
While all hydrothermally prepared n-Cn-PBIs have previously been prepared by conventional methods by others, not all of them had been fully characterized. As a positive side effect, the fact that hydrothermal synthesis allowed us to rapidly synthesize this entire series of n-Cn-PBIs, we could add a considerable amount of to date unreported experimental data on these compounds (see comparison table, ESI†).
Having shown that symmetrically n-alkyl-substituted PBIs can be obtained quantitatively and with great purity within 24 h at 200 °C in H2O, we were interested in studying if: (i) hydrothermal conditions were necessary, or if condensation could also be achieved in H2O at reflux; (ii) If the method was also applicable to other amines (different pKa-values, different steric demands) and (iii) to other bisanhydrides (specifically NBA); and lastly (iv) the completeness and reaction time could be improved adding a non-nucleophilic base as condensation promotor.
To probe if n-Cn-PBIs would be obtainable in H2O at reflux, we took a closer look at 4 PBIs of different polarity and hence solubility of both the monoamine and the resulting PBI: n-C5, n-C11, n-C14 and n-C18-PBI. The conversion to the PBIs was tested by ATR-FTIR, after reaction times of 2, 4, 6, 24 and 72 h. In all cases, the n-Cn-PBIs are forming in considerable amounts, but even after 72 h we still find remaining PBA (ESI†). Therefrom, it can be concluded that HT conditions are indeed required for quantitative yields after reasonable reaction times when equimolar amounts of the starting compounds are employed. Note that Tam-Chang et al. could obtain a perylene monoimide potassium carboxylate, by stirring the parent monoanhydride potassium salt with N,N-diethylethylenediamine in ambient water.14 However, their approach is limited to employing an excess of water-soluble amines in combination with the increased solubility of the monoanhydride potassium salt compared to PBA.
Hydrothermal bisimide syntheses additionally employing Hünig's base (N,N-diisopropylethylamine, HB) as non-nucleophilic base, and/or starting from NBA and/or other amines (cyclohexylamine (c-C6-NH2), aniline (An)) are summarized in Table 1 (ESI† for corresponding ATR-FTIR). First, for n-Cn-PBIs, it becomes clear that the addition of 1–2 drops of HB allows for reducing the reaction time needed for quantitative yield (tR,q) from 24 h (no HB) to 4 h for both n-C8- and n-C14-PBI (Table 1, entries 01–04). However, n-C18-PBI could not be obtained quantitatively by adding HB (Table 1, entry 06). Nonetheless, the consumption of PBA was much improved by the HB addition (ESI,† ATR-FTIR). Second, n-Cn-NBIs were readily obtainable already without the addition of HB: tR,q ≃ 16 h was sufficient to generate n-C8-NBI without HB, which compares to tR,q ≃ 24 h for n-C8-PBI (Table 1, entries 10 and 01). We attribute this to the increased solubility of NBA in H2O in the HT regime compared to PBA. For n-C8-NBI, addition of HB significantly reduced tR,q by a factor of 4, to 4 h (Table 1, entry 11). Most interestingly, n-C18-NBI was obtained quantitatively after tR,q ≃ 16 h, without HB (Table 1, entry 12). Given the high water-insolubility of n-C18-NH2, which did not allow for obtaining n-C18-PBI at any tested conditions, this result is most impressive. Third, cyclohexyl-disubstituted PBI and NBI were obtained quantitatively only in the presence of HB (Table 1, entries 07, 08, 13 and 14), i.e. after only 4 h for c-C6-PBI and 16 h for c-C6-PBI. While the pKa of c-C6-NH2 (≈10.6) is not significantly different from that of n-Cn-NH2 (≈10.4), cyclohexylamine is less nucleophilic due to its steric hindrance.30 This is a possible explanation for the need to add HB for quantitative imidization of both NBA and PBA. Lastly, we tested aniline (An) as monoamine, which is much less basic (pKa ≈ 4.6) than the used alkylamines, and also a weak nucleophile through the mesomeric effect.30 While An-PBI could not be obtained quantitatively after the longest tested tR of 17 h even with HB (Table 1, entry 10), An-NBI was obtained quantitatively without HB after 17 h. This is again most likely related to the higher solubility of NBA compared to PBA, in the HT regime. Clearly, for all employed amines and both bisanhydrides, the speed and completion of the hydrothermal condensation are directly related to the solubility of the starting compounds, their polarity, and nucleophilicity and basicity of the employed amines. Hence, if aiming at N-substituents of the swallow tail or perfluoro-alkyl type – which are of high interest to PBI and NBI dye applications – it is exactly these factors that will have to be considered.
:amine ratio. c-C6-NH2 = cyclohexyl-amine (pKa(c-C6-NH2) ≃ 10.6), An = Aniline (pKa(An) ≃ 4.6), HB = Hünig's base, tR,q = reaction time until bisimides were obtained quantitatively
| Entry | Bisanhydride | Monoamine | HB | t R,q [h] |
|---|---|---|---|---|
| a Not complete after longest tested reaction time. b Considerably higher anhydride consumption than without HB; y = 1–2 drops of HB were added to the reaction. | ||||
| 01 | PBA | n-C8-NH2 | — | 24 |
| 02 | PBA | n-C8-NH2 | y | 4 |
| 03 | PBA | n-C14-NH2 | — | 24 |
| 04 | PBA | n-C14-NH2 | y | 4 |
| 05a | PBA | n-C18-NH2 | — | >24 |
| 06a,b | PBA | n-C18-NH2 | y | >17 |
| 07a | PBA | c-C6-NH2 | — | >24 |
| 08 | PBA | c-C6-NH2 | y | 17 |
| 09a,b | PBA | An | y | >17 |
| 10 | NBA | n-C8-NH2 | — | 16 |
| 11 | NBA | n-C8-NH2 | y | 4 |
| 12 | NBA | n-C18-NH2 | — | 16 |
| 13a | NBA | c-C6-NH2 | — | >17 |
| 14 | NBA | c-C6-NH2 | y | 4 |
| 15 | NBA | An | — | 17 |
In summary, we have prepared a series of high-purity, symmetrically substituted PBIs and NBIs in nothing but water at hydrothermal conditions. The hydrothermal imidization lies in stark contrast to conventional techniques, as: (i) no toxic solvents or catalysts are required at all, (ii) starting compounds were used in stoichiometric amounts, and (iii) no purification (such as recrystallization or chromatography) is required. Moreover, the reactions were complete after reasonable reaction times (max. 24 h) at quantitative yields, with the exception of very hydrophobic amines (n-C16- and n-C18-PBI), and could be accelerated by adding a non-nucleophilic base. Lastly, NBIs are generally more readily formed hydrothermally than PBIs, which we attribute to the increased solubility of NBA in the hydrothermal regime. Our approach presents a rapid and easy means for accessing symmetrically substituted PBIs, which we believe to be of high interest – especially to non-chemists – for the application of these compounds in e.g. energy-related applications.
The authors acknowledge TU Wien for funding this project, and are grateful to Maximilian Raab for preliminary experiments. Powder X-ray diffraction measurements were carried out at the X-ray Center of TU Wien (XRC), and SEM was carried out at the interfaculty electron microscopy facility of TU Wien (USTEM).
References
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed
.
- P. Y. Reddy, S. Kondo, T. Toru and Y. Ueno, J. Org. Chem., 1997, 62, 2652–2654 CrossRef CAS PubMed
- S. S. Ghosh, P. M. Kao, A. W. McCue and H. L. Chappelle, Bioconjugate Chem., 1990, 1, 71–76 CrossRef CAS PubMed
- R. J. Pounder, M. J. Stanford, P. Brooks, S. P. Richards and A. P. Dove, Chem. Commun., 2008, 5158–5160 RSC
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS
- Y. Liao, J. Weber and C. F. Faul, Macromolecules, 2015, 48, 2064–2073 CrossRef CAS
- J. C. Phelan and C. S. P. Sung, Macromolecules, 1997, 30, 6845–6851 CrossRef CAS
- H. Langhals, Heterocycles, 1995, 1, 477–500 CrossRef
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC
- S.-W. Tam-Chang and L. Huang, Chem. Commun., 2008, 1957–1967 RSC
- I. K. Iverson, S. M. Casey, W. Seo, S.-W. Tam-Chang and B. A. Pindzola, Langmuir, 2002, 18, 3510–3516 CrossRef CAS
- S.-W. Tam-Chang, W. Seo and I. K. Iverson, J. Org. Chem., 2004, 69, 2719–2726 CrossRef CAS PubMed
- A. Rademacher, S. Märkle and H. Langhals, Chem. Ber., 1982, 115, 2927–2934 CrossRef CAS
- H. Langhals, Chem. Ber., 1985, 118, 4641–4645 CrossRef CAS
- P. Gawrys, D. Boudinet, M. Zagorska, D. Djurado, J.-M. Verilhac, G. Horowitz, J. Pécaud, S. Pouget and A. Pron, Synth. Met., 2009, 159, 1478–1485 CrossRef CAS
- H. Katz, A. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed
- S. Hünig, J. Groβ, E. F. Lier and H. Quast, Justus Liebigs Ann. Chem., 1973, 1973, 339–358 CrossRef
- M. L. Cheney, G. J. McManus, J. A. Perman, Z. Wang and M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 616–617 CAS
- T. Maki and H. Hashimoto, Bull. Chem. Soc. Jpn., 1954, 27, 602–605 CrossRef CAS
- T. Maki and H. Hashimoto, Bull. Chem. Soc. Jpn., 1952, 25, 411–413 CrossRef CAS
- B. Baumgartner, M. J. Bojdys and M. M. Unterlass, Polym. Chem., 2014, 5, 3771–3776 RSC
- B. Baumgartner, M. J. Bojdys, P. Skrinjar and M. M. Unterlass, Macromol. Chem. Phys., 2016, 217, 485–500 CrossRef CAS
- Y. Nagao and T. Misono, Bull. Chem. Soc. Jpn., 1981, 54, 1191–1194 CrossRef CAS
- H. Huang, Y. Che and L. Zang, Tetrahedron Lett., 2010, 51, 6651–6653 CrossRef CAS
- H. Hall and R. B. Bates, Tetrahedron Lett., 2012, 53, 1830–1832 CrossRef CAS
-
D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa prediction for organic acids and bases, Springer, 1981, vol. 1 Search PubMed
- B. Baumgartner, M. Puchberger and M. M. Unterlass, Polym. Chem., 2015, 6, 5773–5781 RSC
- W. A. Henderson Jr and C. J. Schultz, J. Org. Chem., 1962, 27, 4643–4646 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Materials & methods, ATR-FTIR, SEM, 1H-NMR, 13C-NMR, MALDI-TOF-MS, UV-Vis, fluorescence, PXRD. See DOI: 10.1039/c6cc06567h |
| This journal is © The Royal Society of Chemistry 2017 |