
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnesium, zinc, aluminium and gallium hydride complexes of the transition metals†
Michael J.
Butler
and
Mark R.
Crimmin
 *
*
Department of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: m.crimmin@imperial.ac.uk; Tel: +44 (0)2075942846
First published on 3rd January 2017
Abstract
The preparation and applications of heterobimetallic complexes continue to occupy researchers in the fields of organometallic, main group, and coordination chemistry. This interest stems from the promise these complexes hold as precursors to materials, reagents in synthesis and as new catalysis. Here we survey and organise the state-of-the-art understanding of the TM–H–M linkage (M = Mg, Zn, Al, Ga). We discuss the structure and bonding in these complexes, their known reactivity, and their largely unrealised potential in catalysis.
 Michael J. Butler | Michael J. Butler was born in Tottenham, London in 1989 and in 2012 graduated with an MSci from the University of Glasgow. These studies included a year at Bayer Crop Science AG (Frankfurt am Main), working under Dr Klemens Minn. After some time studying surface chemistry at Sika AG in Zurich, he began an organometallic chemistry PhD in the Crimmin group in 2013. |
 Mark R. Crimmin | Mark R. Crimmin received his PhD in main group chemistry and catalysis from Imperial College London in 2008 supervised by Prof. Mike Hill (now at Bath) and Prof. Tony Barrett. In the same year, he moved to UC Berkeley to study with Prof. Bob Bergman. and Prof. Dean Toste. In 2011, he returned to London as a Royal Society University Research Fellow, initially at UCL and now back at Imperial. Work in the Crimmin group focuses on new methods to break strong carbon–fluorine, carbon–oxygen and carbon–hydrogen bonds in small molecules and the development of heterobimetallic complexes for catalysis. |
1. Introduction
The catalytic practices of C–H bond functionalisation, dehydrocoupling (for hydrogen storage), hydroboration and hydrosilylation are all attractive prospects for the future chemical economy. The modern development in these methodologies continues to be enhanced by the perception of borane and silane σ-complexes as intermediates in reaction mechanisms (Fig. 1).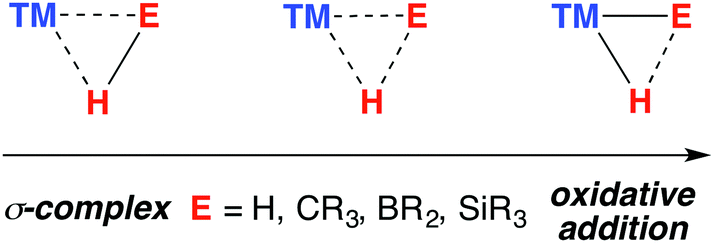 | ||
| Fig. 1 The continuum between σ-complex and oxidative addition for the coordination of E–H bonds to a transition metal. | ||
A σ-complex can be described as an η2-binding of the σ-E–H bond to a transition metal centre (E = C, Si, B, H).1,2 Along with dihydrogen complexes,3–6 σ-silanes are the most comprehensively studied type of this bonding mode.7–12 While the latter appear as potential intermediates in alkene hydrosilylation via the Chalk–Harrod mechanism,13,14 the former bear significance for a range of industrially relevant hydrogenation reactions.15,16 In C–H borylation, stabilisation of catalytic intermediates by a TM–H–B (TM = transition metal) interaction has been supported by significant experimental mechanistic studies.17–19
A 3-centre 2-electron interaction, the η2-ligation of E–H to TM can be viewed within the Dewar–Chatt–Duncanson model and interpreted as a combination of: (i) donation of the σ-electrons in the E–H bond to a vacant orbital on the transition metal and (ii) back-donation from the metal into the σ*-orbital of the same E–H bond. The resulting σ-E–H adduct represents an intermediate along the oxidative addition reaction coordinate (Fig. 1) – part of a continuum of bonding descriptions between free E–H and E–TM–H. It is clear that the nature of the bonding in TM–H–E containing complexes is a tuneable property, being a consideration of the symmetry and energy of the frontier orbitals of the transition metal fragment along with the substituents on, and nature of, E.
A fundamental question that arises when considering this model, is: what happens when C, B and Si are replaced by metallic main group elements such as Mg, Zn, Al, or Ga? The increased ionic contribution within the TM–H–M interaction will necessarily give another dimension to the bonding description. As with silicon, the ability of these elements to expand their coordination sphere leads to the possibility of forming additional bonding interactions with existing ligands on the TM fragment. Furthermore, for the heavier group 13 elements the formation of low-valent MI ligands through manifestation of the inert-pair effect becomes an important consideration.
The TM–H–M motif is one way of adjoining two metal centres bearing at least one reactive hydride ligand (Fig. 2a). This motif can also be obtained by coordinating a transition metal hydride to a neutral main group metal fragment (Fig. 2b), and multiply bridged species formed by a combination of the two aforementioned donor–acceptor interactions (Fig. 2c).
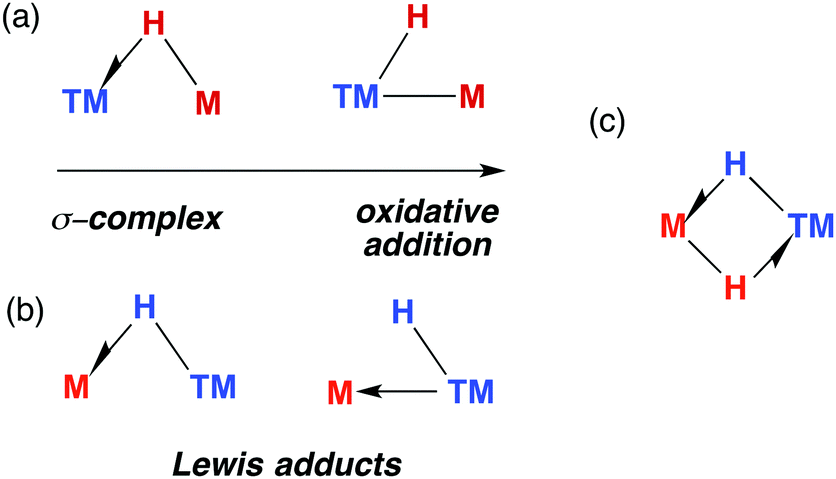 | ||
| Fig. 2 (a) The continuum for addition of M–H to a transition metal. (b) Lewis adducts between a TM hydride and M, (c) combination of interactions. | ||
Herein we survey the known heterobimetallic complexes of transition metal and main group hydrides (M = Mg, Zn, Al, Ga). The coordination chemistry of heterobimetallic transition metal hydrides,20–22 and of Al, Ga, In and Zn-based ligands at transition metal centre have been summarised previously.23–26 To focus the discourse, a loose definition ‘heterobimetallic hydride complexes’ is employed. The complexes mostly fit these criteria: (a) crystallographically characterised; (b) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of transition:main group metal; (c) the absence of TM⋯TM interactions; (d) a hydride ligand in a bridging role. The survey is arranged: TM←H–M, TM–H→M, TM–Hn–M (n > 1).
:1 ratio of transition:main group metal; (c) the absence of TM⋯TM interactions; (d) a hydride ligand in a bridging role. The survey is arranged: TM←H–M, TM–H→M, TM–Hn–M (n > 1).
The half-arrow notation and covalent bond classification advocated by Green, Green and Parkin are used to represent the TM–H–M 3-centre 2-electron interactions.11 This formalism is employed as an organisational principle not as an absolute interpretation of the bonding within TM–H–M units. The line drawings are constructed from the perspective of the transition metal coordination environment in order to represent charge neutral species rather than an accurate representation of the bonding within the TM–H–M group. The literature survey is followed by discussion of the “continuum” of bonding descriptions, reactivity and the potential these complexes hold for catalysis.
2. σ-Complexes (TM←H–M)
We have reported 1, a Zn congener of structurally related σ-alane complexes (Fig. 3).27 The binding of the zinc hydride to the CuI centre is weak and reversible. In toluene or benzene solution, an equilibrium exists between the heterobimetallic complex and the η2-arene complex of CuI. The electronic structure of the three-centre linkage has been investigated by DFT calculations. The analysis suggests that the formal L donation of the M–H σ-bond to the 4s orbital on CuI is accompanied by weak Cu→M back-donation into the M–H σ*-orbital (see Discussion section). In combination, the data allow 1 to be described as a weakly bound σ-complex.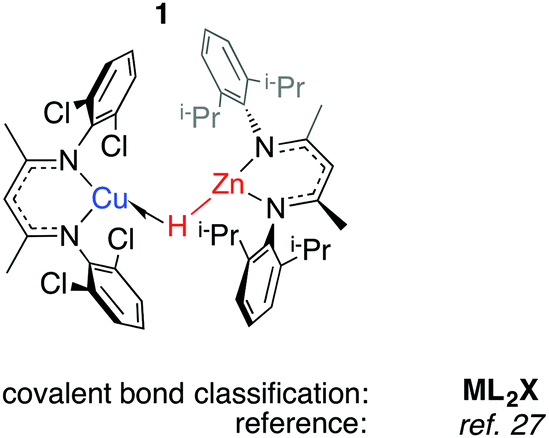 | ||
| Fig. 3 A σ-zincane complex of CuI. | ||
Coordination of Al–H bonds to CuI has also been examined; complexes possessing four-coordinate and five-coordinate aluminium centres have been isolated (Fig. 4, 2 and 3). The binding of Cu–H–Al is again weak and reversible based on solution NMR studies and crossover experiments. In DFT calculations, the coordination of exogenous ligands to the CuI fragment was found be increasingly exergonic across the series C6F6 < H–B < H–Si < C6H6 < H–Zn < H–Al – a trend that is manifest in the experimental data.27
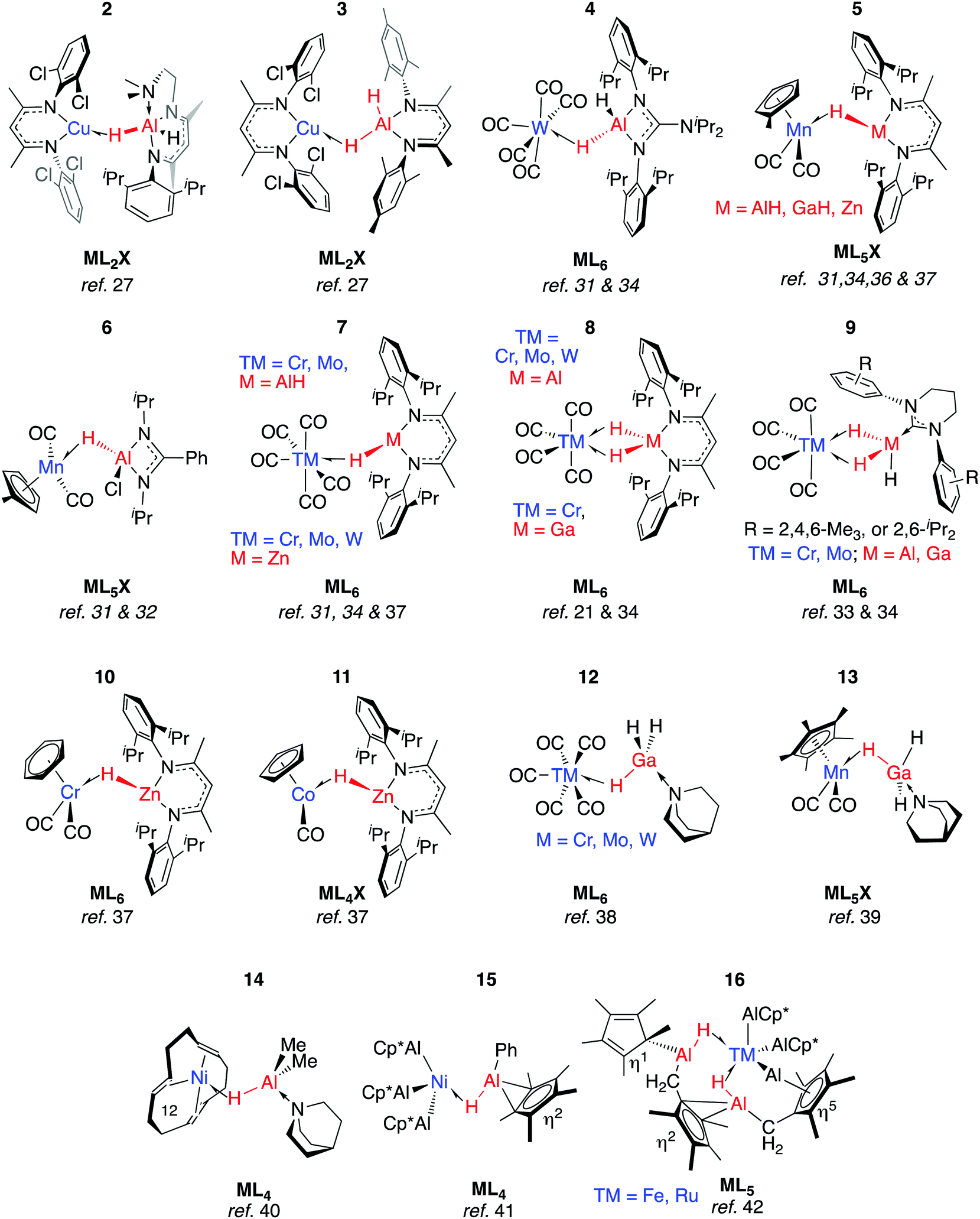 | ||
| Fig. 4 σ-Alane, σ-gallane and σ-zincane complexes of late TM. | ||
In 2 the Cu⋯Al vector lies outside of the H–Al–H wedge – the only example of this structural feature we are aware of in main group metal σ-complexes.27 The ‘lee side’ coordination of the Al–H bond and very long intermetallic distance in 2 are notable. For comparison, σ-complexes of HBpin (pinacolborane) or HBcat (catecholborane) show different coordination geometries to those of four-coordinate boranes BH3·EMe3 (E = N, P).28,29 The discrepancy has been rationalised by disruption of the TM⋯B back-donation due to the absence of a vacant orbital of suitable energy in BH3·EMe3, be it the boron p-orbital or the σ*-orbital of the B–H bond.28 While steric factors are undoubtedly important, a similar effect may explain the solid state structures of 2 and 3. DFT calculations are consistent with reduced back-donation from d10 CuI into the Al–H σ*-orbital of the five-coordinate species when compared to the four-coordinate analogue.27 Higher nuclearity species containing Cu–H–Al interactions are known and the intermetallic cluster [(Cp*AlCu)6H4] has been obtained from reaction of [Cp*Al]4 with [Ph3PCuH]6.30
Aldridge and co-workers pioneered this area of research and have isolated a series of σ-alane and σ-gallane complexes of groups 6 and 7 transition metal carbonyls.31–35 Like 2 and 3, the structures of 4–7 contain σ-Al–H adducts and derive from ligand exchange reactions, in this case from the parent metal carbonyl under either thermal or photochemical conditions. Four-electron, η2:η2-coordination of H–Al–H (8–9) requires a 14-electron transition metal fragment, {M(CO)4} and necessitates displacement of two ligands from ML6 starting materials.31,33,36 Complexes 7-Cr/Al and 8-Cr/Al have not been separated and, like their heavier congeners, 7-Mo/Al and 8-Mo/Al were isolated in an approximate 9:1 mixture with the minor component containing the η2:η2-coordination mode.34 The adduct 8-W/Al could not be obtained by photoejection of CO from [W(CO)6] and was finally obtained under thermal conditions using [W(CO)4(1,5-COD)] as a precursor (COD = cyclooctadiene).34 The M⋯Al distance in 8-W/Al is shorter than would be expected based on the lighter members of the series, presumably due to a tighter binding of the σ-alane to the more expanded 5d orbitals of W. In contrast to the W analogue, 8-Cr/Al may be formed directly upon heating 7-Cr/Al: Eyring analysis and the first order kinetics of this reaction have led Aldridge and co-workers to suggest it proceeds by an associative pathway.34
All TM–H–Al heterobimetallic complexes characterised by Aldridge and co-workers show slow exchange between the bridging and terminal hydride ligands on the NMR timescale at ambient temperature.31–34,36 From structural and spectroscopic evaluation of these complexes (4–9), it appears that back-donation into the M–H σ* orbital is negligible. While the close TM⋯M contacts (especially in 9-Ga) are short enough to hint at TM–M interaction, the four-membered ring imparted by the η2:η2-coordination mode in 8–9 demands such a short contact.33 The weaker nature of the Ga–H bonds (cf. Al–H) means 8-Ga is only a minor product of the reaction of the corresponding gallane with [M(CO)4(1,5-COD)] as this species is unstable with respect to dihydrogen elimination (vide infra).36
Although the coordination chemistry of Zn–H–TM and Mg–H–TM groups remains underdeveloped when compared to the aluminium analogues, we have recently reported a series of σ-zincane complexes of closely related transition metal carbonyl fragments. Ligand exchange reactions readily occur under photochemical conditions and 5-Zn, 7-Zn, 10–11 have been isolated and crystallographically characterised.37
Ueno and co-workers provided evidence that the η2:η2-coordination mode is not necessary for the stabilisation of σ-gallanes, and reported 12 and 13.38,39 These complexes were formed from displacement of THF or CO from group 6 transition metal carbonyls by GaH3·quinuclidine. While there is little account for TM⋯Ga interactions in 12, 12-W again contains a shorter TM⋯Ga separation than what would be reasoned from inspection of the solid state structures 12-Cr and 12-Mo.38,39 Complex 13 is noticeably similar to 6 in terms of the TM–H–M parameters. In both cases, the Cpcentroid–Mn–H–M torsion angle is near 90° allowing for overlap of the M–H σ*-orbital with the HOMO of the Mn fragment. There are also similarities to the first reported example of a σ-alane complex. Formally a Ni0 species possessing a triene ligand and a coordinated alane, 14 contains an unsupported Ni–Al–H linkage that survives the substitution of the tridentate ligand with three equivalents of CO to form [(CO)3Ni(μ-H)AlMe2·quinuclidine].40
An alternative approach to TM–H–Al groups has been discovered by Fischer and co-workers. Addition of [Cp*Al]4 to transition metals with labile ligands is proposed to generate intermediates of the form [TM(AlCp*)n] which react further, effecting the inter- or intramolecular C–H activation of arenes or alkanes.41,42 Complexes 15–16 are formed through this route and possess geometries that are consistent with the σ-alanes described above (Fig. 4). For example, the Al–H distance in 15 of 1.76(3) Å lies within the range established σ-alanes. While 16 possesses elongated Al–H distances ranging from 1.88(8) to 1.89(7) Å, these are still substantially shorter than the >2.0 Å separation required to suggest oxidative addition (vide infra). The latter may be described as a complex containing stretched σ-alane ligands with an Al–H–TM geometry somewhere between coordination and oxidative addition.
3. Oxidative addition/hydride transfer (H–TM–M)
We have reported the thermal reaction of M–H bonds (M = Al, Zn, Mg) with [Cp*Rh(H)2(SiEt3)2] to form 17 and 18,43 a reaction that is believed to proceed through the 16-electron intermediate {Cp*RhH(SiEt3)}. These species are different to the σ-complexes described above and data are consistent with the product of oxidative addition (Fig. 5).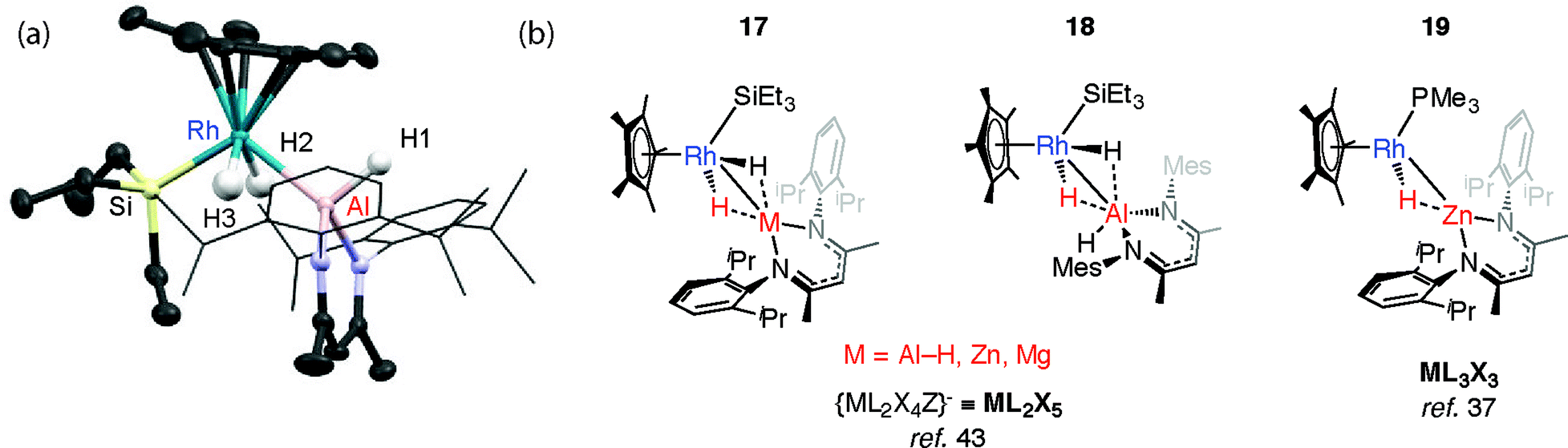 | ||
| Fig. 5 (a) The crystal structure of 17-Al. Selected bond lengths (Å): Rh(1)–Si(1) 2.3668(7), Rh(1)–Al(1) 2.4579(7), Al(1)–H(1) 1.51(2), Al(1)–H(2) 2.13(3), Al(1)–H(3) 2.22(3), Rh–H(2), 1.47(3), Rh–H(3) 1.53(3). (b) Oxidative addition (hydride transfer) of M–H bonds to a Rh complex. | ||
In all cases the TM–M distances are within the sum of covalent radii and the M⋯H distances stretch to well beyond 2.0 Å. Moreover the four-legged piano-stool geometry around the Rh centre, including the trans-relation of the hydride ligands, is conserved when compared with silane and borane analogues, [Cp*Rh(H)2(SiEt3)(X)] (X = SiEt3, Bpin). The description of 18 as an oxidative addition product is supported by the significant 1JRh–H value of 40.2 Hz and terminal ν(Rh–H) frequency of 1966 cm−1. The calculated charges (NBO analysis) on Rh are significant for only the Zn and Mg analogues. As such, while these Zn and Mg complexes could be described as oxidative addition products, hydride transfer to form an is also a fair description.43
The structure of 17-Al, an analogue of 18 that incorporates more sterically demanding substituents on the β-diketiminate ligand, shows a geometry with familiar trans-disposed hydrides, short TM–Al distance and long Al⋯H distances (Fig. 5). Reaction of 17-Zn with an excess of PMe3 under photochemical conditions leads to the elimination of an equivalent of silane and formation of 19. This latter heterobimetallic complex again contains a short TM–M distance and a M⋯H separation of greater than >2.1 Å.37
Mindiola and co-workers have isolated a related Fe–H–Mg complex, albeit as a minor component of a mixture formed upon reaction of EtMgCl with an iron chloride precursor.44 Complex 20 also results from C–H activation of the ligand (Fig. 6).45 While it could be assigned as an σ-Mg–H complex of Fe the long Mg⋯H distance and short Fe–H separation make this debatable, it is arguably closer to an ate-complex formed by a tightly bound Fe–H−→Mg+ ion-pair. Although both 20 and 17–19 can be described as the products of hydride transfer the key difference is that the latter are formed from addition of the M–H bond to the TM centre and contain a defined and quantifiable TM–M bond.
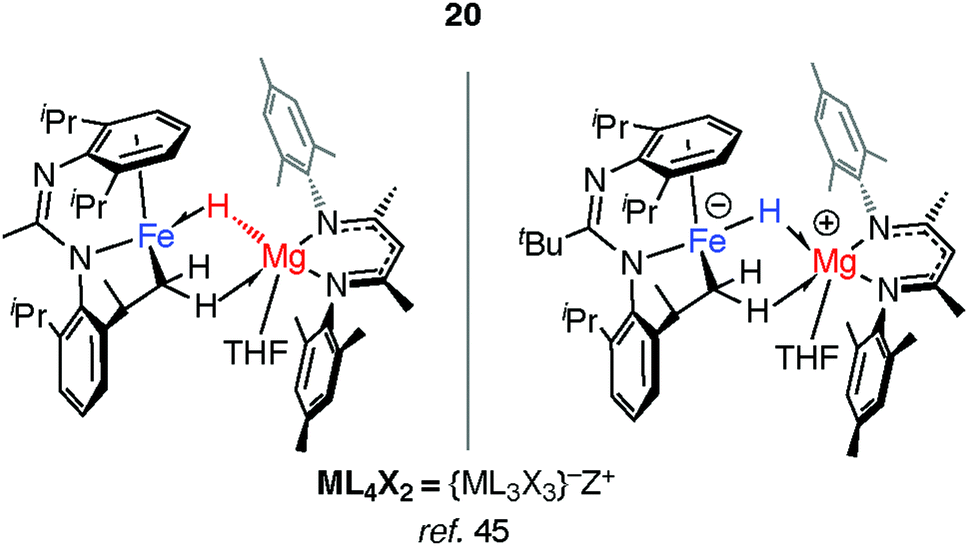 | ||
| Fig. 6 Hydride transfer to form a tightly bound ion-pair in 20. | ||
4. Low-valent main group ligands from dehydrogenation (TM←M)
The products of oxidative addition of M–H bonds to transition metals may only be intermediates on a path to coordinated low-valent fragments (M = Al, Ga). As the main group is descended, not only does the reduced M–H bond strength result in easier M–H “bond activation”, but the manifestation of the inert pair effect means the lower common oxidation state becomes increasingly stable.Two examples of the generation of AlI ligands by the dehydrogenation of aluminium dihydride precursors in the coordination sphere of a transition metal have been reported. The products of these reactions retain an undeniable H⋯Al interaction. The photochemical elimination of HSiEt3 from 19 leads to the dimer 21 (Fig. 7).43 Complex 21 contains a Rh2Al2H4 core. Supporting the argument for AlI is the deviation of the alumocycle from planarity, suggestive of decreased π-donation by the N atoms into the 3p orbital of Al. This allows for AlI to act as both a Z- and L-type ligand with respect to Rh.43 Structurally related intermetallic compounds containing Pt2Ga2H4,46 Ru2GaH2,47 Ru2Ga2H4,48 Ru2Al2H4,48 Co2H2Al2,49 Rh2Zn2H2 groups are all known,50,51 as are higher nuclearity species in which multiple main group fragments act as ligands for the transition metal (see ESI,† Fig. S1).52–57 The work on cluster complexes supported by organozinc, organoaluminum and organogallium ligands has been reviewed before.23–26
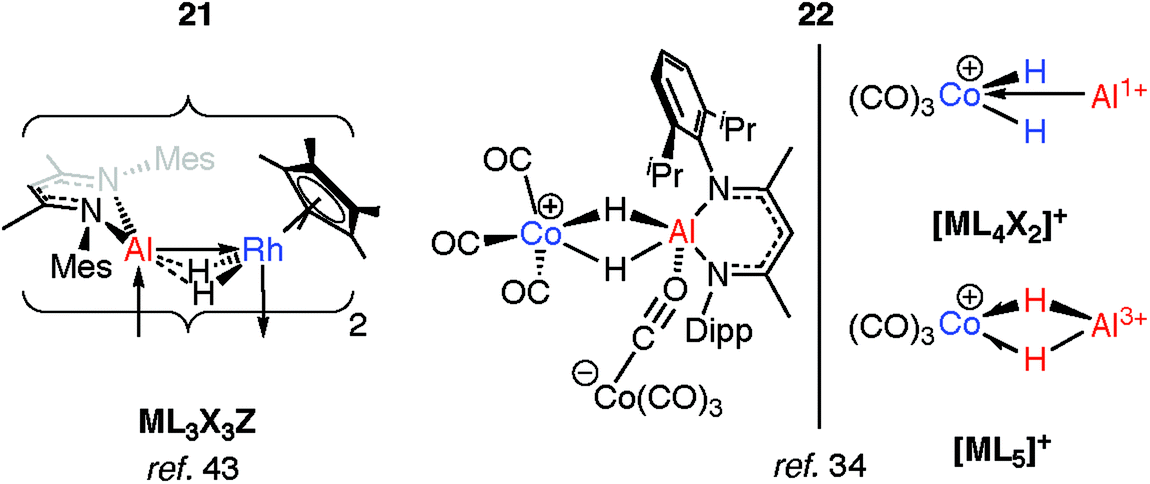 | ||
| Fig. 7 Generation of AlI ligands from addition of aluminium dihydrides to TM. | ||
Coordination of a related aluminium dihydride to a 14-electron {CoI(CO)3}+ synthon gives 22 (Fig. 7).34 This latter species appears to be a product of simultaneous addition of both Al–H bonds to Co. The bridging Al⋯H distances of 1.92(3)–1.98(3) Å in 22 are not as long as those found in the oxidative addition products 17-Al or 18 of 2.0–2.2 Å but are significantly longer than those found in σ-complexes of the same aluminium species which typically range from 1.6–1.8 Å. As with 21, the aluminium centre receives additional electron density, here by end-on coordination of an isocarbonyl ligand of the formally anionic {Co(CO)4} moiety. The electronic structure of 22 lies somewhere between the bis σ-complex and the dehydrogenated CoIII/AlI species (Fig. 7).
Complexation of gallanes to late transition metal carbonyls (TM = Cr, Mo, W, Mn, Fe, Co) has been shown to lead to the formation of GaI ligands through spontaneous or photoinduced extrusion of dihydrogen (Fig. 8).34,36 Complexes 23 and 24 are formed from addition of gallanes to [(η5-C5H4Me)Mn(CO)3] and [TM(CO)n] (TM = Cr, Mo, W, n = 6; TM = Fe, n = 5) precursors. A bimetallic reaction intermediate is proposed, from which 1,2-elimination of dihydrogen occurs. Reactions employing 34-electron carbonyls [Mn2(CO)10] or [Co2(CO)8] in place of the 18-electron [M(CO)n] complexes proceed similarly. For Co, oxidative addition is followed by H2 elimination across the TM–M bond to give 25. For Mn, oxidative addition of the Ga–H bond is followed by reductive elimination of H–TM(CO)4 and α-migration of the remaining hydride from Ga to Mn to form 26 (see Discussion, Fig. 23).36 The proposed intermediate of the latter reaction contains a Mn–Ga–H group and finds experimental support from the work of the groups of both Driess and Fischer who have reported complexes containing Fe←Ga–H,58 and TM←M–H moieties (TM = Cr, Mo, Zn; M = Al, Ga).59–61
 | ||
| Fig. 8 Generation of GaI ligands from addition of gallium dihydrides to TM. | ||
5. Hydride-bridged Lewis adducts (TM–H→M)
Late transition metal adducts
Lewis acidic main group metal centres without hydride substituents may be coordinated by transition metal hydrides in two different ways: (i) through a direct metal–metal interaction from donation of d-electrons of the TM to the main group metal (Z-type ligand) or (ii) through 3-centre 2-electron hydride bridges of the form TM–H→M (Fig. 2). Elimination reactions may result from these adducts provided the pKa of the hydride is low enough and the metal alkyl (or aryl) basic enough to effect alkane (or arene) elimination.For example, Andersen and Bergman have reported the reaction of [Cp*Ir(H)2(PMe3)] with a series of organo-aluminium and -magnesium compounds (Fig. 9).62 Coordination of Ph3Al to form 27 results in the widening of the H–Ir–H angle by 20° and signifies dominant TM–M bonding. In contrast, the reaction of [Cp*Ir(H)2(PMe3)] with Ph2Mg(THF)2 eliminates benzene and forms 28, while that with AlEt3 eliminates two equiv. of ethane and yields the dimeric species [Cp*IrPMe3(μ-AlEt)]2, 29. Both 28 and 29 activate CO2, giving [Cp*Ir(PMe3)CO] as the TM-containing product.62
 | ||
| Fig. 9 Late transition metal adducts. | ||
The TM⋯M distances in 27–29 are in range of the sum of the covalent radii, and the coordination at iridium in 27 is such that the Ir–Al bond is distorted away from the IrH2 plane by 37°.62 The geometries contrast with those of 30-W and 30-Mo. First reported by Wailes et al.63 and Storr et al.,6430-W has been characterised by X-ray diffraction and analysed by DFT methods.65,66 While the current understanding is that the two metals interact through a single hydride bridge to Al, there is limited data to support a non-negligible TM→M donation. Firstly, the HOMO on the TM fragment is calculated to be a metal-localised d-orbital-type suitable for electron donation. Secondly, the BX3 (X = F, Cl) adducts of the same TM fragments were calculated with the boron atom lying outside of the H–TM–H wedge.66
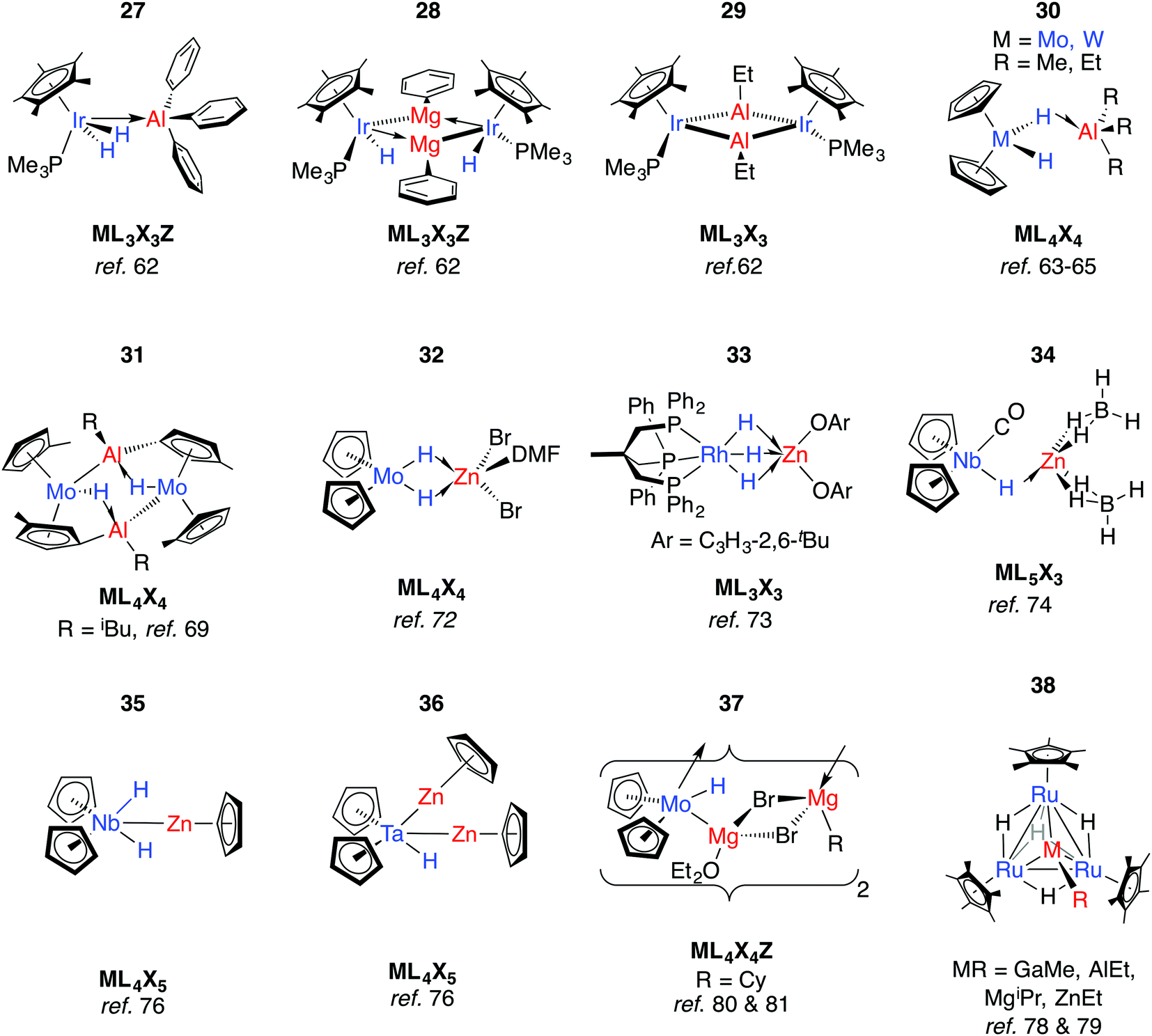
Although the gallium and boron analogues of 30 are yet to be isolated, 1H NMR data for a GaMe3/[Cp2WH2] admixture shows likely equilibrium between free compounds and a weakly bound adduct.67 These data are consistent with the known acceptor strength of the Lewis acids AlMe3 > GaMe3 > BMe3. Rhenium analogues of 30 were discovered by Wailes et al. using the metallocene [Cp2ReH].63 The potential for reversible H/D exchange between the hydrides and protons of the cyclopentadienyl ring of 30-W has been highlighted and proposed to occur by a mechanism involving anchimeric assistance.65 Non-reversible intramolecular deprotonation of the cyclopentadienyl ligands is also well established, and often leads to high nuclearity species such as complex 31.68–71
When [Cp2MoH2] was treated with ethylzinc bromide, 32 was isolated and presumed to derive from ZnBr2 formed from a Schlenk-type equilibrium.72 An η2:η2-bonding mode of the Mo hydrides is implied by the narrowing of the MoH2 wedge upon adduct formation, data that contrast those of 27. A similar adduct, 33, is formed from addition of a bis-aryloxy zinc solvate to a Rh trihydride complex.73
Reaction of a mixture of NbCl5, sodium cyclopentadienyl and zinc powder in THF under an atmosphere of CO, and subsequent treatment with NaBH4 gives 34.74 The metal⋯metal distance in 34 is just within the sum of the covalent radii and there is a shift of the ν(CO) absorption upon coordination of the Lewis acid to [Cp2Nb(CO)H] (1960 to 1910 cm−1). Both findings mark a significant Nb → Zn interaction. In contrast, only a small shift in the ν(CO) absorption is seen upon reaction of [Cp2NbH(CO)] with AlEt3.75 Based on significant changes to the hydride resonance observed by 1H NMR data, the coordination of AlEt3 in [Cp2Nb(L)H·AlEt3] (L = CO, C2H2, PMe3) is proposed to occur through a μ-hydride ligand rather than a direct metal⋯metal interaction. Related reactions between a 1:1 mixture of [Cp2NbH3] or [Cp2TaH3] and [Cp2Zn] do not lead to adduct formation but cyclopentadiene elimination and formation of new species 35 and 36 both of which contain TM–Zn bonds.76,77 Tebbe and co-workers reported the d1 complex [(Cp2NbH2)2Zn] from the 1:2 reaction of Et2Zn with [Cp2NbH3].75 Higher nuclearity species 36–38 have been isolated from reactions of ruthenium polyhydride complexes or [Cp2MoH2] with main group alkyls (Fig. 9).78–81
In more recent work, Bourissou, Uhl and co-workers have shown that hydrogenation of an intramolecularly coordinated Pt→Al adduct leads to the heterobimetallic hydride 39 (Fig. 10).82 Calculations suggest that H2 addition occurs across the Pt→Al bond. There is precedent for this intramolecular coordination mode: Fischer and co-workers have reported the gallium adduct 40.83 Related hydrogenation reactions of platinium diene complexes either bearing a Z-type Al ligand or in the presence of GaI co-ligands lead to the formation of heterobimetallic complexes bearing terminal hydride ligands on the transition metal (Fig. S1, ESI†).46,84 For example, 41 is formed upon hydrogenation of a Pt⋯norbornadiene precursor; it is currently unclear if the Pt→Al moiety plays a role in dihydrogen activation (Fig. 10).84
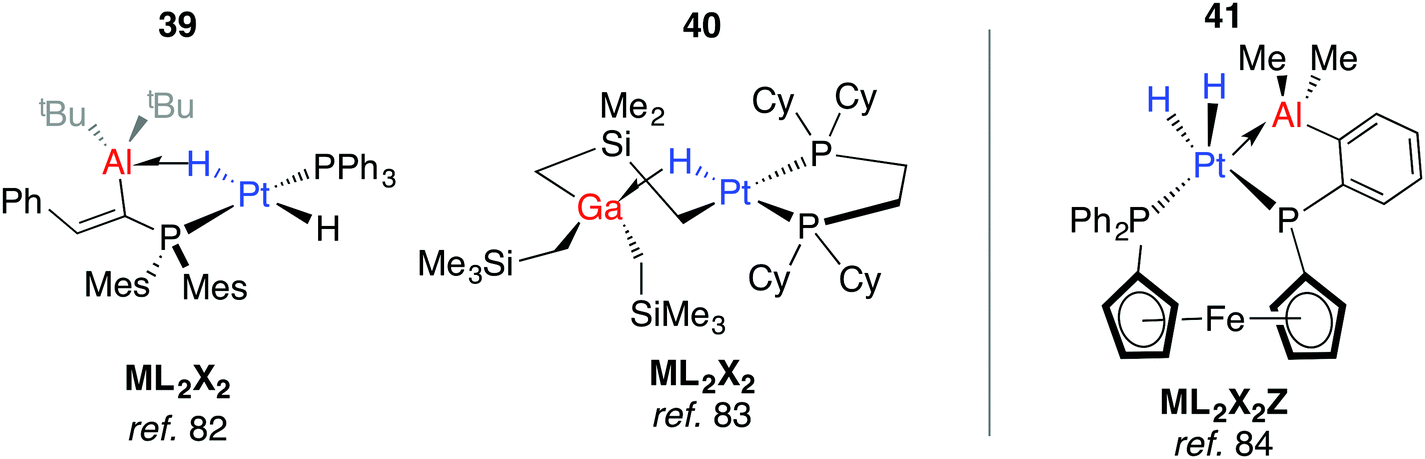 | ||
| Fig. 10 Adducts from H2 or C–H addition across a Pt→Al Bond. | ||
Early transition metal adducts
Relevant to Ziegler–Natta polymerisation, Zr–H→Al Lewis acid–base interactions have been known for some time.85 Coordinatively saturated, d0 heterobimetallic complexes may be formed by coordination of the parent hydride to a Lewis acid. For example, 42 was suggested by Wailes and co-workers based on 1H NMR experiments as early as 1972.67 Crystallographic characterisation of related adducts 43 and 44 containing inter- and intramolecular Zr–H–Al moieties was later reported.86,87 While there is less data for hafnium, the adduct 45 has been reported and is notable for the location of the Al centre outside of the H–Hf–H wedge. The position of the hydrides are unusual when compared to related group 4 metallocenes as is the 16-electron configuration of Hf.88 For group five analogues, [Cp2TaH3·MEtn] forms irreversibly for M = Al, Ga (n = 3) but reversibly for Zn (n = 2).75 Solid state infrared spectroscopy and X-ray diffraction data for a related complex, 46, are consistent with formation of an adduct containing two Ta–H–Zn linkages forming an η2:η2-coordination mode, although NMR data suggest that only one bridging hydride is retained in solution (Fig. 11).89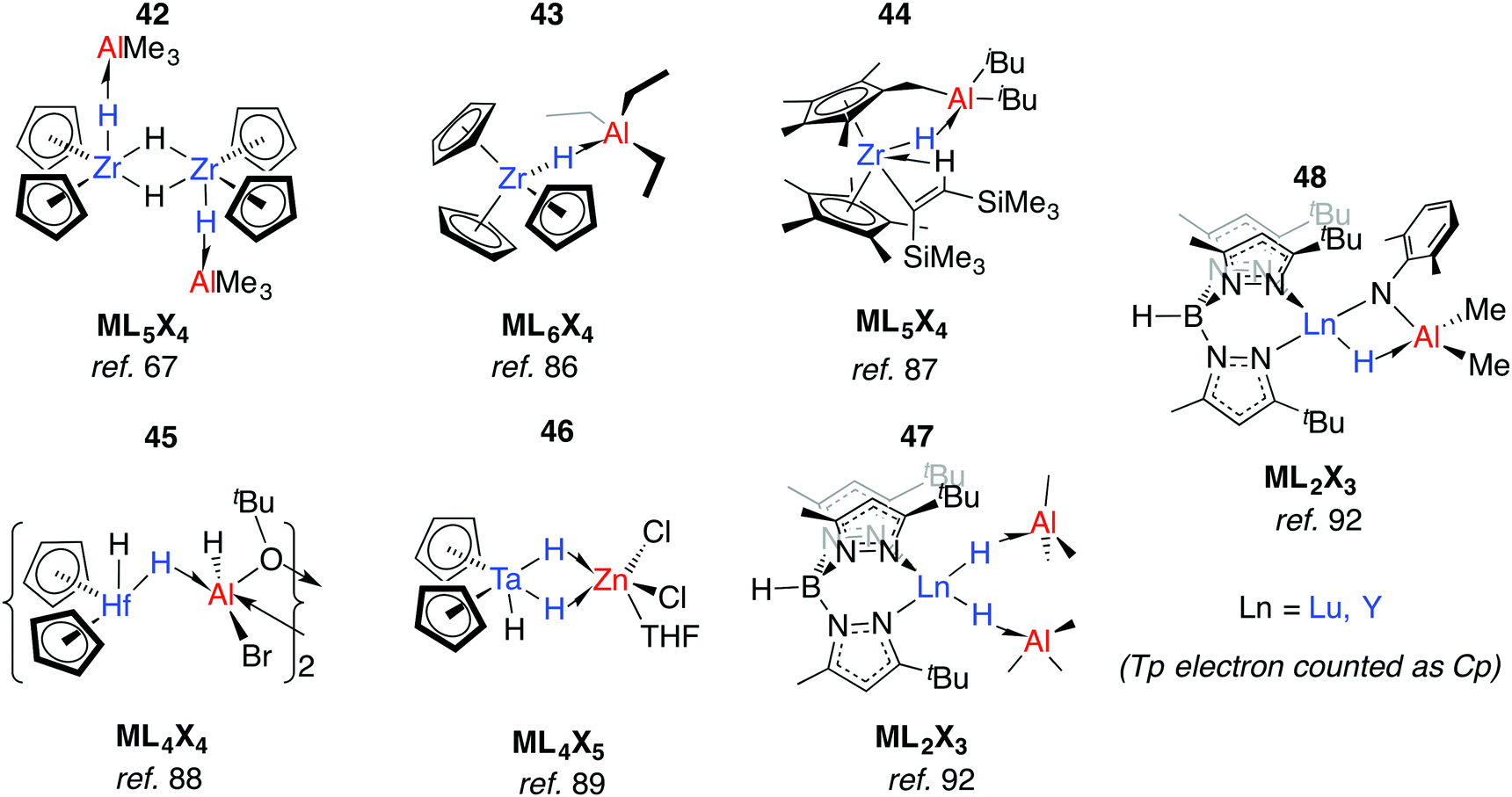 | ||
| Fig. 11 Early TM/main group adducts. | ||
Anwander et al. have investigated the reactions of yttrium and lutetium alkyl and amide complexes with aluminium hydrides.90–93 For example, lanthanide hydride bonds in 47 are capped and stabilised by Lewis acidic aluminium sites.92 This motif is not unique to 48, and X-ray data has been collected on monomeric [Cp2Lu(μ-H)·AlH3·NEt3],94 and dimeric [Cp*Y(Me)(μ-H)AlMe2(μ-H)]2.95 Despite the coordinated Lewis acid, 47 displays reactivity consistent with a terminal hydride; deprotonation of 2,6-dimethylaniline gave complex 48. Here, the imide linker supports an intramolecular Ln–H→Al interaction (Fig. 11).92
6. Multiply-bridged complexes (TM–Hn–M, n > 1)
Rare earth metal adducts
[(C5Me5)2YMe(THF)] reacts with HAl{N(SiMe3)2}2 allowing trapping of μ-H2-bridged 49.96 The alane in this complex is strongly bound and addition of donor, including chelating, ligands could not effect separation of the heterobimetallic complex. The coordination mode in 49 is predated by those found in 50–55 reported by Bulychev and co-workers in the 1980s.94,97–105 In these latter complexes, the Al fragment and hydride source derive from either LiAlH4 or AlH3·L (L = NEt3, THF, OEt2), and either the chloride or hydride ligands take up μ3-coordination modes. Amongst the complexes [Cp2YH·AlH3L]2 (50), the Y⋯Al distances decrease, and Al–H stretching frequencies increase, across the series L = Et2O < THF < NEt3. The five-coordinate Al centres found in these complexes are notably different to the borate analogues, where the boron atom does not engage in coordination numbers higher than 4 and solvation occurs at the rare earth metal centre (Fig. 12).106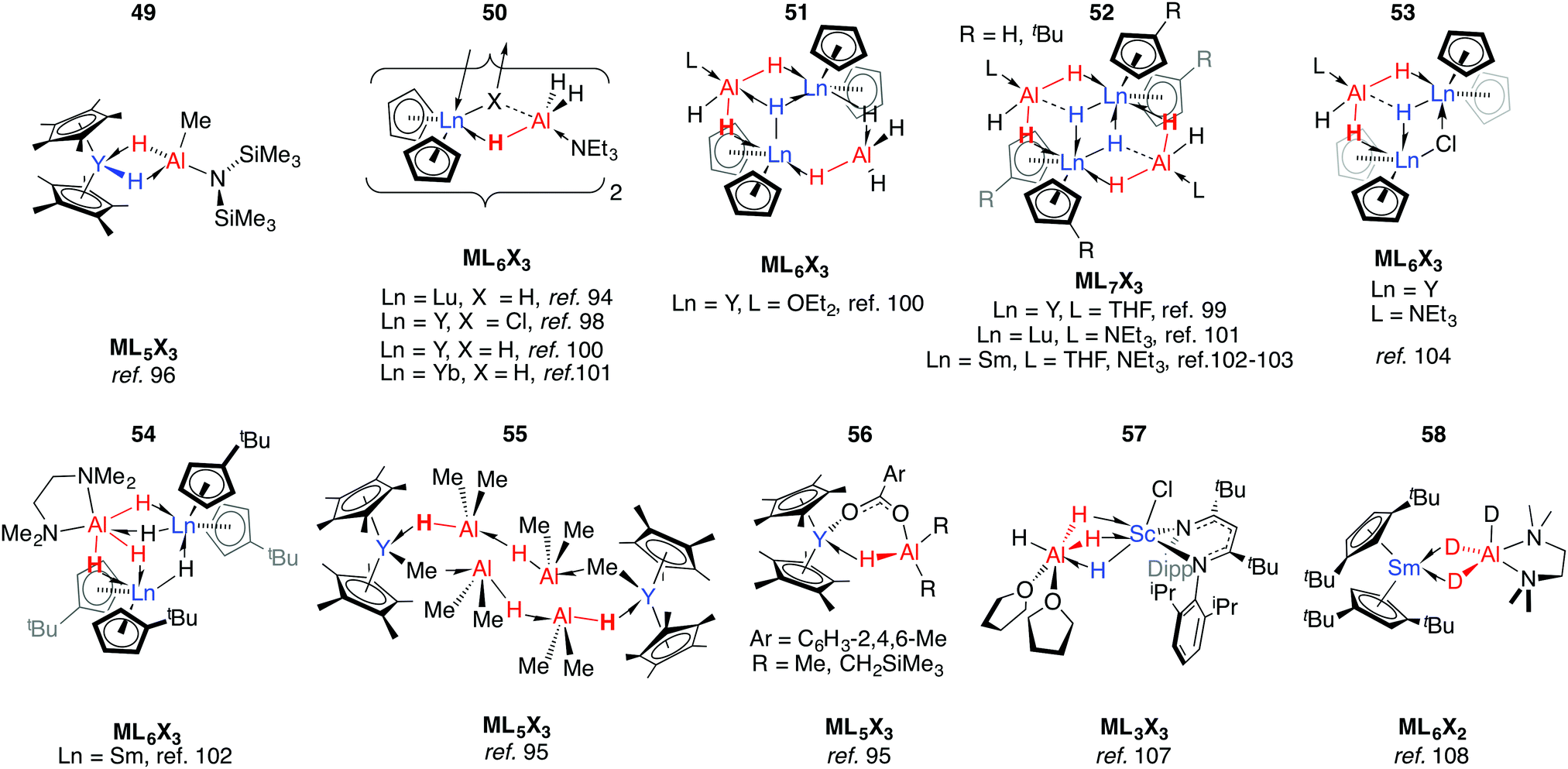 | ||
| Fig. 12 Coordination of aluminium hydrides to rare earth metal centres. | ||
Alanes react with yttrocene hydride or carboxylate complexes to form Y←H–Al adducts (55–56).95 While investigating the salt metathesis of a β-diketiminato-supported Sc dichloride complex with LiAlH4, Piers and co-workers isolated the alane adduct, 57.107 In 57, both metals are six coordinate, and this is the only time a (μ-H)3 bridging motif seen in the absence of a late TM. Heterobimetallic hydrides of rare earth metals are not limited to those in which the heavy metal is in the 3+ oxidation. Bulychev et al. found that [(1,3-tBu2-C5H3)2Sm] partially decomposes into an octanuclear aggregate of SmIII and alane, when treated with AlH3 in the presence of TMEDA. Upon substitution of the alane for AlD3, however, 58 may be isolated: an apparent effect of isotopic substitution.108
Group 4 metal adducts
As part of understanding the role of methylaluminoxane in polymerization catalysis,109 Bercaw, Britzinger and others have reported NMR studies on hydride-bridged Zr/Al oligomers, trimers and dimers in solution (59–60).85,110–112 They concluded that binuclear structures of type 60 are only produced for ansa-metallocenes and inclusion of a terminal Me ligand on Zr causes decomposition. Cationic Zr- and Hf-analogues are known.113–115 It has been shown that {Zr(μ-H)3Al2}+ reacts reversibly with AlMe3 or ClAliBu2 to give dimethyl- or dichloro-bridged Zr/Al species (Fig. 13).114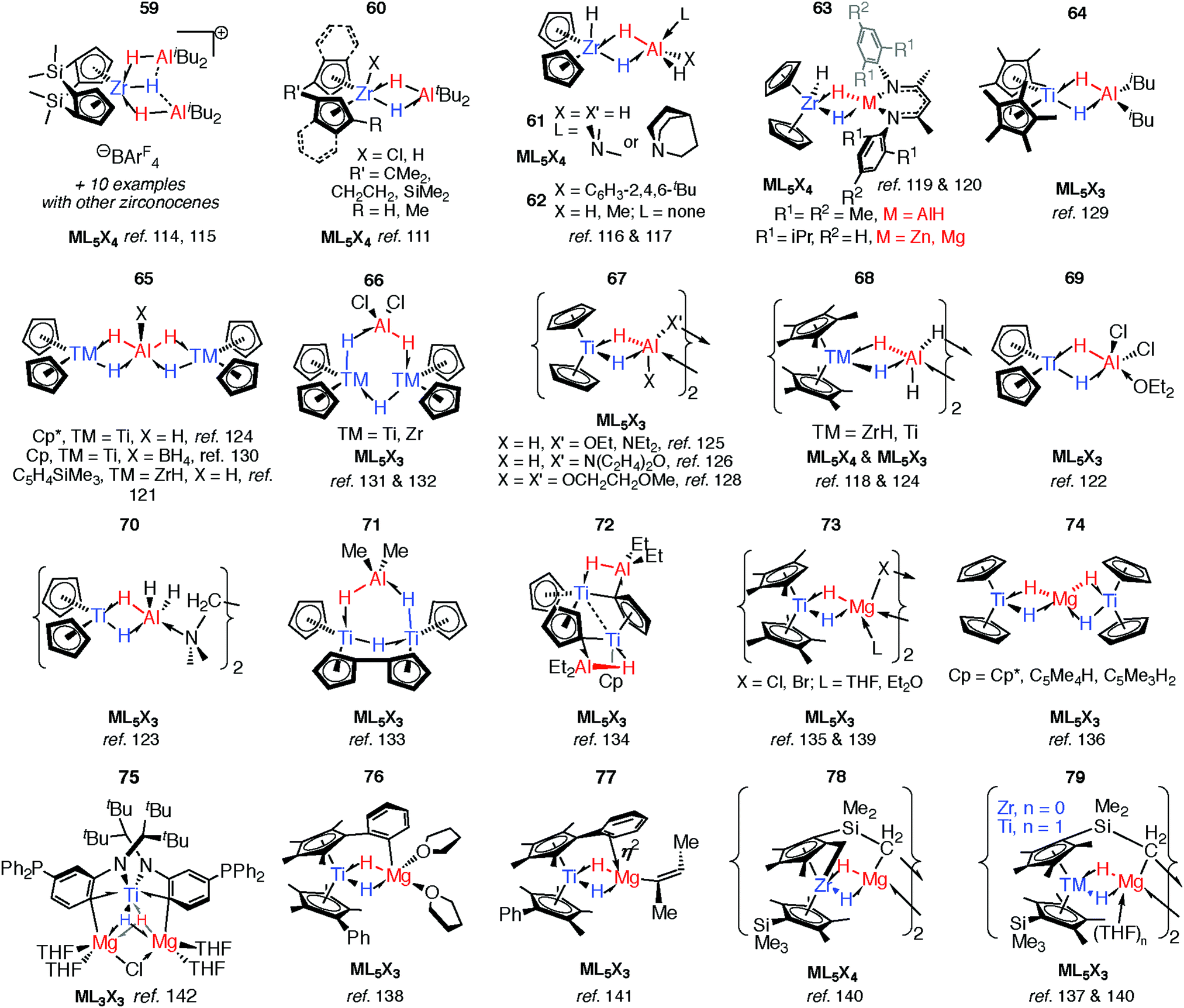 | ||
| Fig. 13 Group 4 complexes with multiple hydride bridges. | ||
An early example of a Zr–H–Al complex was published in 1997 by Raston and co-workers.116 Structurally and synthetically, 61 is logical extension of the work of Bulychev et al. (vide supra). These species are poorly soluble in all common organic solvents but THF, and require stabilisation by a base. Power and Wehmschulte added a super bulky aryl group (62) improving solubility and eradicating the need for base stabilisation.117 Base-free complexes have also been reported by Stephan and colleagues.118 NMR data and calculations for the monomers 62 suggest both fast intramolecular hydride exchange and an intermolecular exchange between the heterobimetallic complex and its homometallic parts in solution. Our group has also published an example of this type: soluble in toluene and hexane 63-Al exists in equilibrium with the alane and [Cp2ZrH(μ-H)]2.119 In contrast, the homologues, 63-Mg and 63-Zn show no sign of dissociation into monometallic parts. This may be a result of tighter binding due to an increased ionic contribution to the donor–acceptor linkage (Fig. 13).120
Many of the ZrIV/Al heterobimetallic hydride complexes are synthetically accessible by salt-metathesis reactions of the parent zirconocene dihalide with aluminium hydride reagents.121 Similar reactions with titanium(IV) precursors commonly lead to isolation of products with Ti in the 3+ oxidation state.122–128 This is displayed most nakedly in the recent report of 64 by Beweries.129 Six-, five- and four- coordinate aluminium centres have been reported in {Ti2Al2H8}, {Ti2AlH5}, and {TiAlH2} cores respectively (65–72).118,121–134 There is only limited precedent for similar reduction chemistry occurring with Zr.131,132 The observed coordination modes of AlxHy units in these and related group 4 complexes are common to heterobimetallics and are found in related Ta, Nb, Mn and Ru complexes (vide infra).
Substitution of the terminal hydride ligands on aluminium for halide, alkyl, alkoxide or amide has little influence on structure.133,134 Magnesium-based heterobimetallic complexes of group 4 metallocenes (73–77) can be prepared by reaction of the parent metallocene dichloride with either Grignard reagents or Mg0 powder in etheric solvents.135–142 For preparations in which a main group hydride is not used as a reagent the hydride ligands result from either: (i) C–H atom abstraction from the solvent (THF), (ii) β-hydride elimination group derived from a main group alkyl, or (iii) C–H bond activation of the cyclopentadienyl ligand (or its substituents). Ligand activation is common in these complexes and intramolecular deprotonation to form dianionic Cp ligands, including “tuck-in” complexes has been observed to lead to both diamagnetic TMIV and paramagnetic TiIII complexes (72 & 78–79).126,133,134,143,144
Reaction of 63-Zn with an excess of 1,5-cyclooctadiene resulted in the on-metal transformation of the organic diene to an alkyne adduct, 80 (Fig. 14).120 This is an example of a heterobimetallic complex containing a planar, four-coordinate carbon and is related to the first confirmed zirconium-ethylene complex 81, reported by Parkin and co-workers.145 Structurally related zirconium(IV) and titanium(IV) species are known in which the TM–H–M connection is supported by bridging metallacyclopropane or metallacyclopropene ligands (80–88, Fig. 14).86,120,146–153 A ketene analogue is also known which incorporates an oxycyclopropane ring.154 The structures of 84–87 are distorted to accommodate additional intramolecular binding interactions. Calculations have shown that electron donation from the bridging Zr–C bond to the Lewis acidic main group centre plays a key role in the stabilisation of these species (82) and explains the unusual geometry at the bridging carbon.147 A combination of computational methods (NBO and QTAIM) have led our group to describe the bridging ligand of 80 as a slipped metallocyclopropene.120
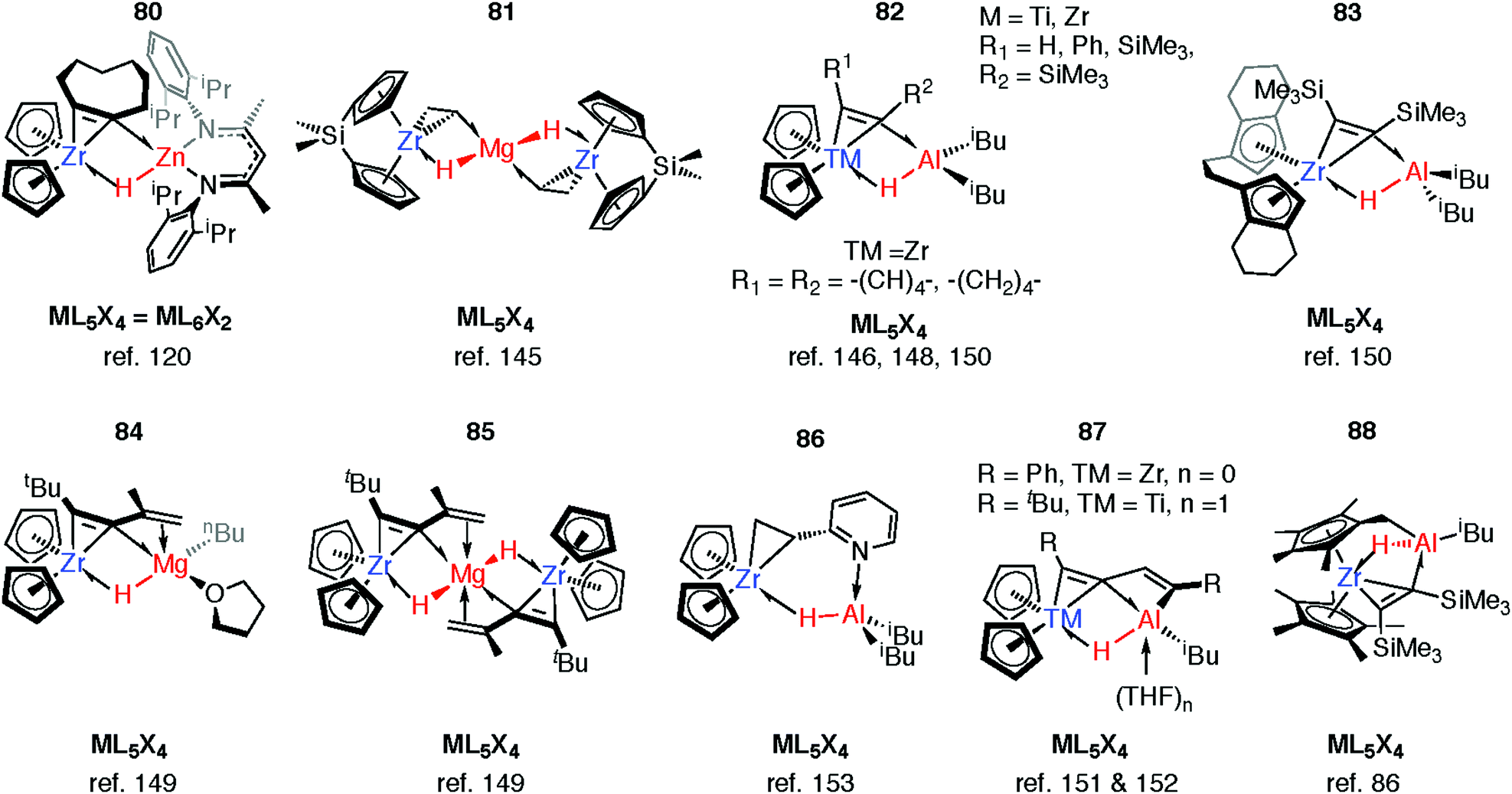 | ||
| Fig. 14 Coordination of M–H to Zr and Ti metallocyclopropene and metallocyclopropane complexes. | ||
Late transition metal adducts
Multiply-bridged main group complexes (M = Mg, Zn, Al and Ga) of late transition metals have been known for more than 50 years. Much of the early work is limited by the accuracy of the spectroscopic methods of the period.20 Nevertheless, it has been clear for some time that the electronegativity difference between late TM and main group metals creates a significant ionic contribution to the bonding.20 Complexes 89–97 are mostly products of salt metathesis between lithium aluminohydrides and TM chlorides (Fig. 15).155–171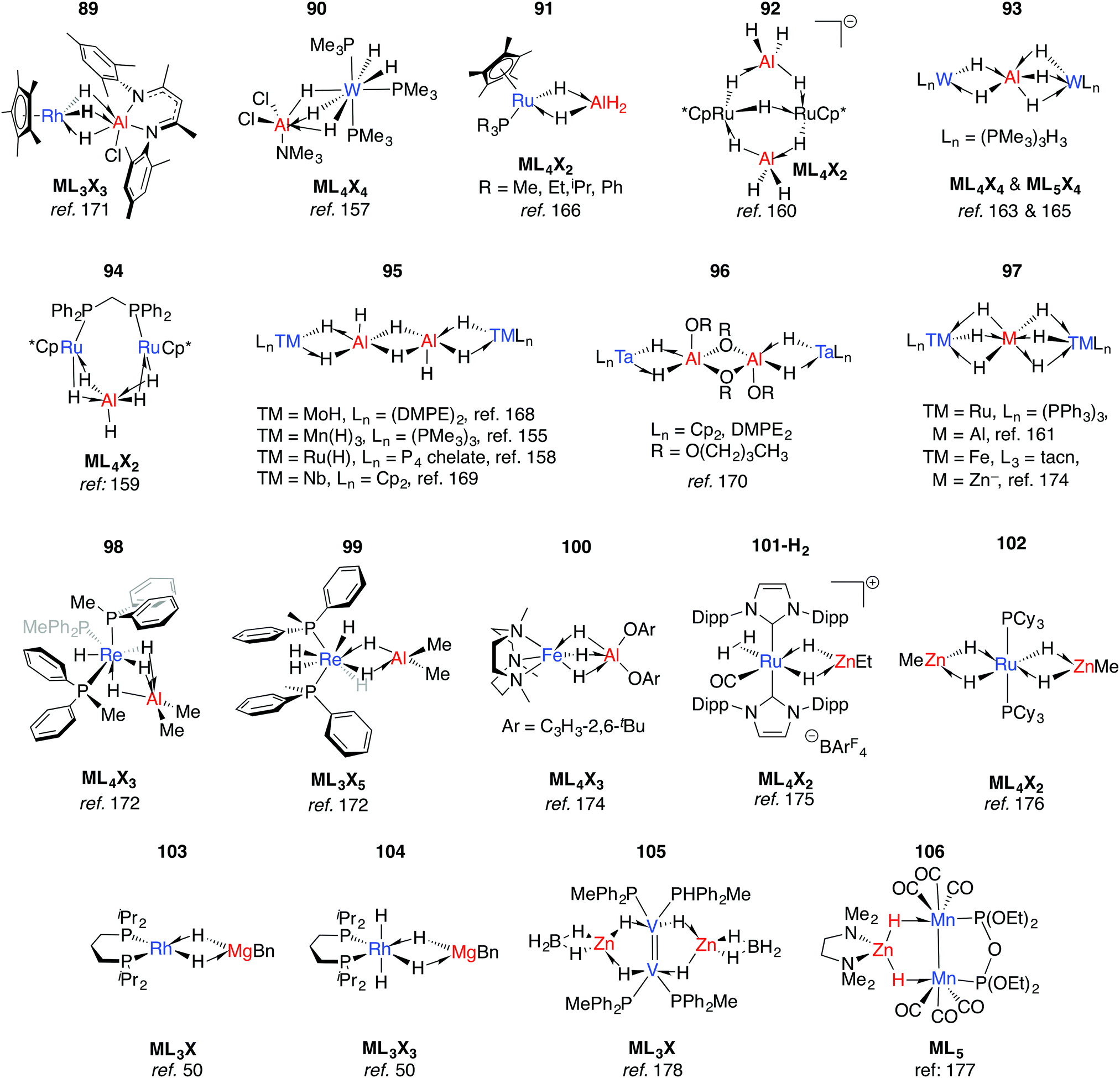 | ||
| Fig. 15 Late TM heterobimetallics containing multiple bridging hydrides (tacn = 1,4,7-triazacyclononane). | ||
Common themes emerge in the coordination geometries of these complexes.170 Unlike the group 4 analogues described above, the (μ-H)3 bridge is a reoccurring feature. Geometries at aluminium are typically trigonal pyramidal (5-coordinate)159 or distorted-octahedral (6-coordinate). The high coordination number and the geometry of ligands around the TM centres provides coordinative saturation, and in most cases makes the existence of a metal–metal bond very unlikely. For 89, this finding is in direct contrast to the data reported for the related Rh/Al species 17-Al and 18 (Fig. 5).171
In solution many of these complexes are fluxional and provide time-averaged 1H NMR chemical shifts for bridging and terminal hydrides. In relation to this phenomenon, the complexes 98 and 99 reillustrate the ionic component of the bonding. The main group fragment of 98 and 99 is {AlMe2}+, which may exchange between different positions on the face of the {P3ReH4}− and {P2ReH6}− polyhedra.172 For comparison, [ReH9]2− is well-known as the dipotassium salt.173
The low-spin ground state structure of 100 has been thoroughly investigated by computational methods.174 The frontier molecular orbitals of 100 do not support bonding between Fe and Al, and the authors present the complex as being dominated by a strong donation of electron density from a {Fe–H}− fragment to {AlX2}+ (X = O-C6H3-2,6-tBu). This conclusion is a good general insight into the electronic structure of the late-TM(H)nAl bonding, the dimeric examples of which are summarised in 95–96.
An alternative synthetic approach to multiply-bridged main group complexes of the late transition metals is through the addition of H2 across an unsupported Ru–Zn bond. The Ru(H)2Zn moiety in 101-H2 discovered by Whittlesey and co-workers demonstrated good thermodynamic stability and survives extrusion of dihydrogen by heating in vacuo to form 101.175 The hydride ligand trans to H2 in 101-H2 is protic and significantly closer to Ru, while the hydride trans to CO is hydridic and equidistant between the metals. NBO calculations show no sign of Ru–Zn interaction in either complex. However, a QTAIM calculation on 101 shows a Ru–Zn bond path and only one Zn–H bond. It can tentatively be concluded that the μ-H atoms in 101-H2 and 101 are intermediate and “flexible” between terminal and bridging character. Fischer and co-workers have reported the related complex 102, which results from addition of 4.5 equivalents of ZnMe2 to a Ru/Al heterometallic complex prepared in situ by mixing [Ru(PCy3)2(η2-H2)(H)2] and [Cp*Al]4.176 Two decades before, Rh/Mg complexes 103 and 104 were prepared by Fryzuk, however their thermal instability precluded characterisation by anything other than 1H and 31P NMR spectroscopy (Fig. 15). A number of dinuclear (with respect to TM) complexes supported by chelating ligands or involving TM–TM bonding including 105–106 are also known.177,178
7. Discussion
An exhaustive account of the preparation and structures of heterobimetallic hydride complexes reported over the last half a century is presented above. During our own research in this area and through analysis of the material above, we have alighted upon recurring issues in the understanding this family of complexes. For the benefit of potential future studies, these issues are discussed below.(i) Structure and bonding
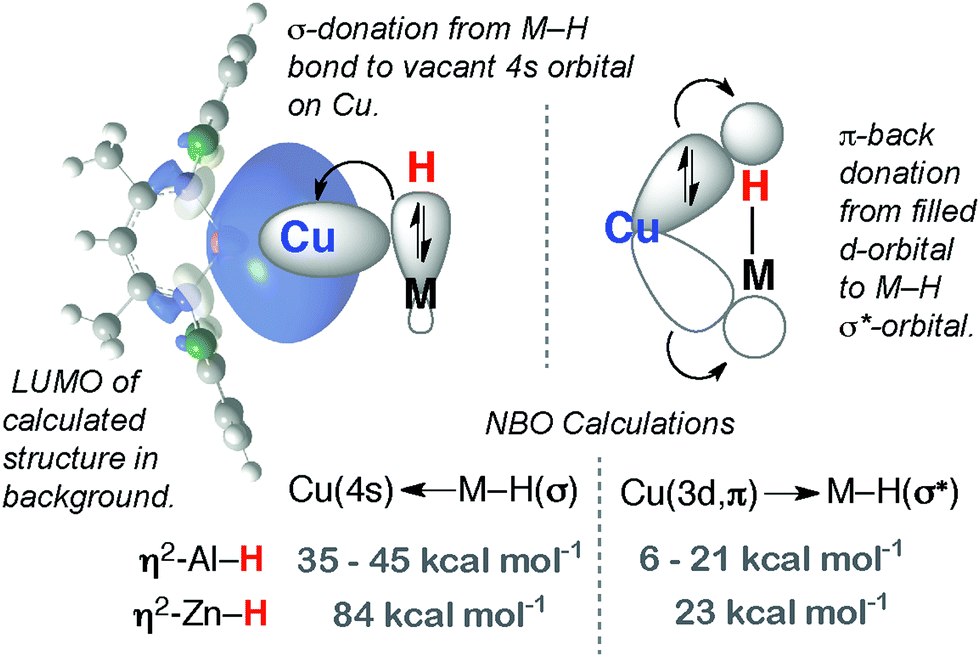 | ||
| Fig. 16 Bonding description of 1–3. | ||
The model is supported by comparison of the carbonyl stretching frequencies of complexes of the form [(η5-C5H4Me)Mn(CO)2L] where L is an E–H or M–H bond. Listed in Table 1 these data show that σ-alane, σ-gallane and σ-zincane complexes are similar to those formed from four coordinate boranes such as H3B·NMe3. The data are consistent with limited back bonding into the M–H bond and contrast those of three coordinate σ-boranes and σ-silanes where increased back-donation to the E–H bond results in higher frequency carbonyl stretching frequencies.37 The domination of the σ-donation component of the bonding, and the importance of the ionic component to bonding, is readily understood by considering the difference in the Pauling electronegativity of the elements involved for coordination of E–H (Δχp: B = 0.18, Si = 0.32) and M–H bonds (Δχp: Mg = 0.92, Zn = 0.57, Al = 0.61, Ga = 0.41).
| L | ν 1/cm−1 | ν 2/cm−1 |
|---|---|---|
| H2 | 1982 | 1922 |
| HBCat | 1995 | 1937 |
| HSiPh3 | 1983 | 1926 |
| HGePh3 | 1965 | 1910 |
| HSnPh3 | 1934 | 1925 |
| H3B·NMe3 | 1918 | 1839 |
| H–Al(H)BDI | 1947 | 1879 |
| H–Ga(H)BDI | 1951 | 1886 |
| H–ZnBDI | 1937 | 1852 |
Considering the structures of the isolated complexes in Sections 2 and 3, it becomes clear that the well understood continuum between σ-complexes and oxidative addition products detailed for addition of E–H bonds to transition metals also applies to M–H bonds.
The triangular TM–H–M unit is subject to structural changes as the electron density changes at the TM (Fig. 17). The formal shortness ratio (fsr) normalises the metal⋯metal distance and has been used to evaluate the intermetallic interaction in complexes containing two metals in close proximity. For data collected to date, this metric appears to conveniently describe the extremes of the reaction coordinate: σ-complexes (fsr approx. >1) and products of oxidative addition/hydride transfer (fsr approx. ≤1).179 One caveat of this approach is that short metal⋯metal distances can arise due to the geometric constraints imposed by multiple bridging ligands, to date the analysis has only been applied to molecules containing a single TM–H–M unit.
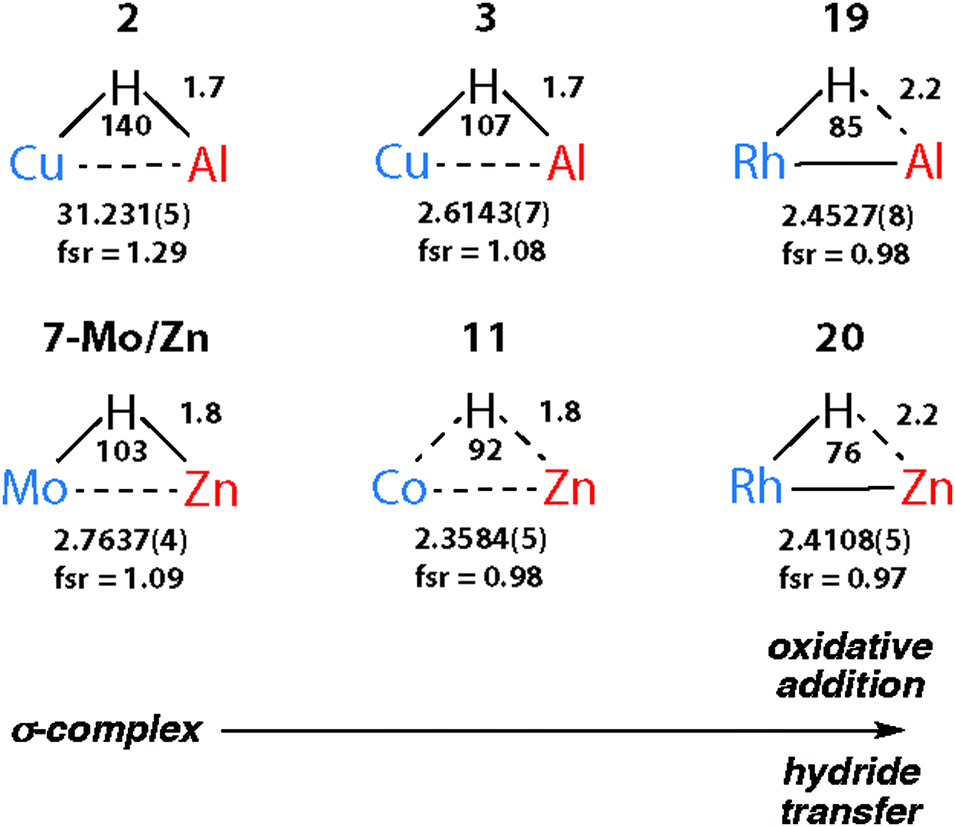 | ||
| Fig. 17 Comparison of key distances (Å) and angles (°) from X-ray (TM⋯M) structures of coordinated Al–H and Zn–H bonds. Trends confirmed by DFT studies. | ||
Very recently we have described the reaction coordinate for the addition of zinc-hydrides to transition metal centres.37 Through isolation of a series of different transition metal complexes and DFT studies we outlined a continuum which is characterised by the transfer of electron density from the breaking largely ionic Zn–H bond to forming, increasingly covalent, TM–H and TM–Zn bonds (Fig. 18). Only at very short TM⋯Zn separations does the Zn–H bond begin to lengthen significantly. The Zn–H bond varies between approx. 1.7–1.8 Å for σ-zincane complexes but increases dramatically to 2.2 Å for the product of oxidative addition. Similarly for σ-alane complexes the Al–H separation of 1.6–1.8 Å increases to >2.0 Å for products defined as oxidative addition. The breaking point of this bond appears to occur quite late along the reaction coordinate. While high quality and low temperature X-ray data is becoming increasingly well established for the location of hydride ligands in solid state structures, there is clearly significant error in the TM–H and M–H distances and associated TM–H–M angle. In the case of the analysis presented in Fig. 18, this concern is circumvented by performing DFT calculations to confirm the location of the hydride.
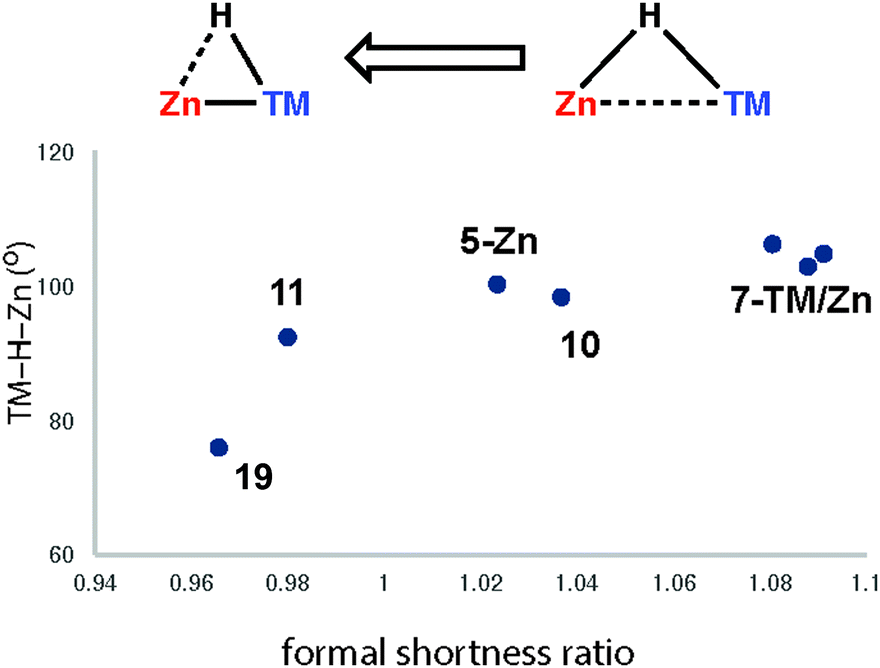 | ||
| Fig. 18 Reaction coordinate for the approach of a Zn–H bond to a transition metal. Adapted from ref. 37. | ||
Comparison of a series of E–H and M–H complexes of a single transition metal fragment, {Cp*Rh(H)(SiEt3)} has allowed us to conclude that the ionic component to bonding remains important for the products of oxidative addition and is such that for M = Zn and Mg these complexes can accurately be described as a result of hydride transfer. The two extreme valence bond description of 17 and the related H–SiEt3 and H–Bpin complexes are represented in Fig. 19. DFT calculations show that the covalent contribution to the TM–M bond (Wiberg Bond Index) decreases along the series Mg ∼ Zn < Al < Si ∼ B while the charge on rhodium becomes significant for the Zn and Mg members of the series (NPA charge on Rh: Bpin = −0.03, SiEt3 =−0.06, Al(H)BDI = −0.11, ZnBDI = −1.01, MgBDI = −0.99).43 In combination the data suggest that the ionic contribution becomes more significant for the more electropositive elements.
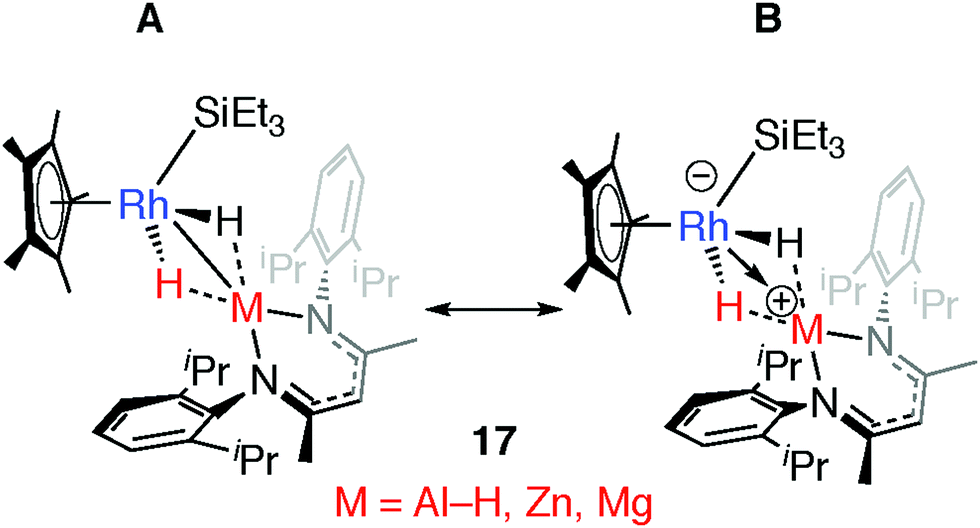 | ||
| Fig. 19 Extreme bonding description of 17 and related complexes. Adapted from ref. 43. | ||
Hydride transfer can also occur to generate complexes that do not possess a TM–M bond such as the tight ion-pair 20.45 In the extreme case, charge separation could occur to form a completely ionic species, such as the salt [Zn(NH3)4][Cr2(μ-H)(CO)10]2.180
There are clear parallels between the zwitterionic valence bond description B and the neutral donor–acceptor complexes, TM→M–H. These species are connected parts of a continuum of bonding descriptions as are σ-complexes of the form TM←H–M and TM–H→M adducts. It appears that as the fsr provides some insight when comparing coordination complexes of the M–H bond to TM, it may also be of some use in considering coordination of the TM–H fragment to M. The bond distances and angles around the Nb–H–Zn moiety of 34 and 35 are compared in Fig. 20. The fsr ratio decreases and TM–H–M angle becomes increasingly acute when comparing a complex with a genuine TM–M bond (34) against that in which the primary interaction between the two metals occurs through a 3-centre 2-electron TM–H–M bond.
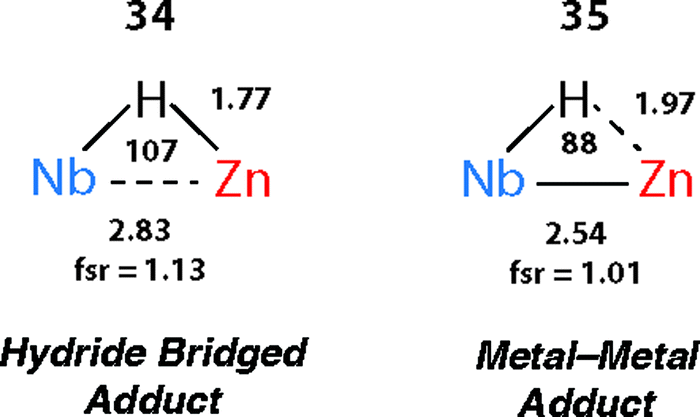 | ||
| Fig. 20 Comparison of the bond lengths (Å) and angles (°) in 34 and 35. Figure adapted from ref. 76. | ||
While, in general, the higher electronegativity of the main group element in silanes (and to an extent boranes) means that TM–H→ERn bonding descriptions are less common, there is growing appreciation that these descriptions may be relevant in σ-silane and σ-borane chemistry. For example, an Ir–H−→SiR3+ interaction has recently been invoked to explain the electronic structure of an intermediate isolated during catalytic studies in the dehydrocoupling of silanes and alcohols.181 Related borane-capped transition metal hydrides, especially those generated by reversible addition across TM→B bonds are gaining increasing attention in catalytic hydrogenation processes.182
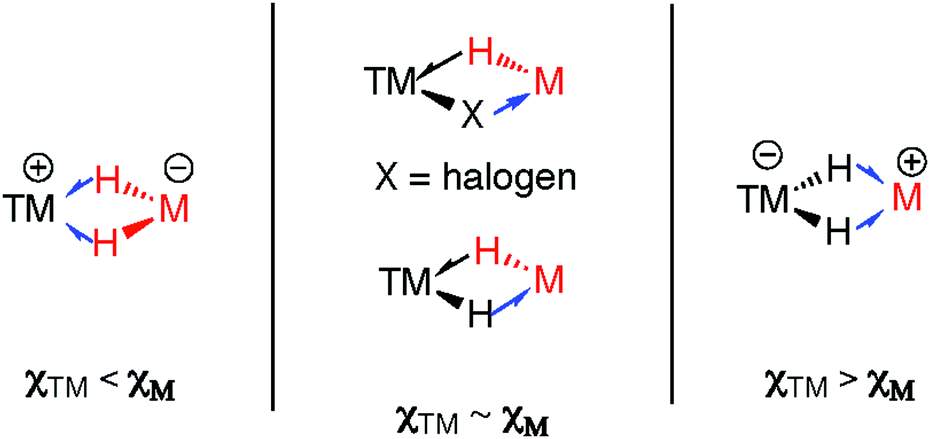 | ||
| Fig. 21 Limiting bonding descriptions in multiply bridged heterobimetallic complexes. | ||
A number of species have been isolated that can be considered coordination complexes of the trapped parent hydrides, these include MgH42−, AlH4−, AlH52−, AlH62−, ZnH64−, and Al2H82− the structural cores of which can be compared to similar complexes containing SiH4, SiH62− trapped between two transition metals.183,184 These species elucidate the general coordination modes found in multiply hydride bridged species (Fig. 22). More recently, we have found that the aggregation state of the complexes may be reduced by incorporating kinetically stabilising ligands on M.27,37,43,119,120,171
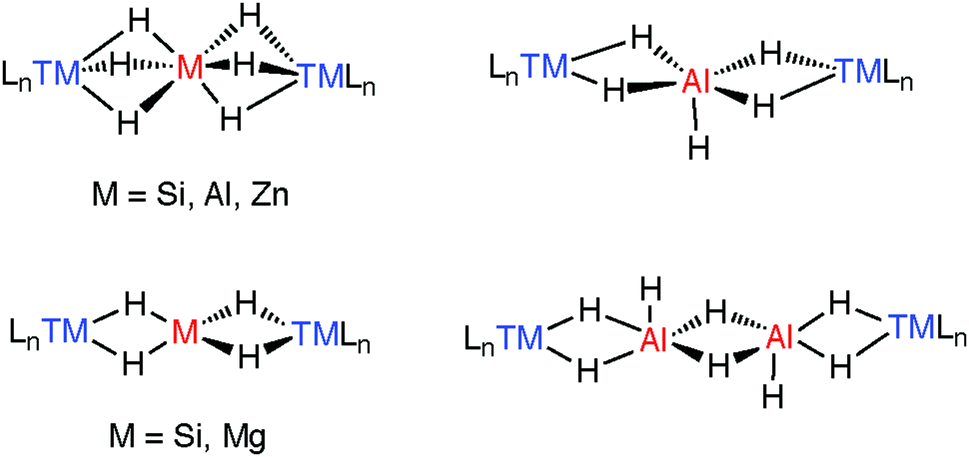 | ||
| Fig. 22 Coordination geometries of trapped MHxn− units and comparison to silane chemistry. | ||
(ii) Reactivity
The 3-centre 2-elecron TM–H–M moiety is surprisingly stable. The novel ground state structures found in heterobimetallic TM–H–M complexes are often assumed to herald unusual reactivity. This correlation is far from proven. Indeed, when it comes to reactivity, the understanding of the ground state structure is not enough. For example, a common feature of the data for the multiply-bridged hydrides is exchange between hydride positions in the solution phase.175By far the most well understood reaction of TM–H–M groups are those which involve elimination of dihydrogen. Experimentally, while this reactivity has been observed for both Al and Ga complexes, examples of the latter dominate. Through the isolation of intermediates and kinetic analysis, a number of mechanisms for H2 elimination have been identified and are presented in Fig. 23. Both α-migration and 1,2-elimination of H2 across a TM–M bond have been used to rationalise the formation of GaI ligands in the coordination sphere of transition metals.
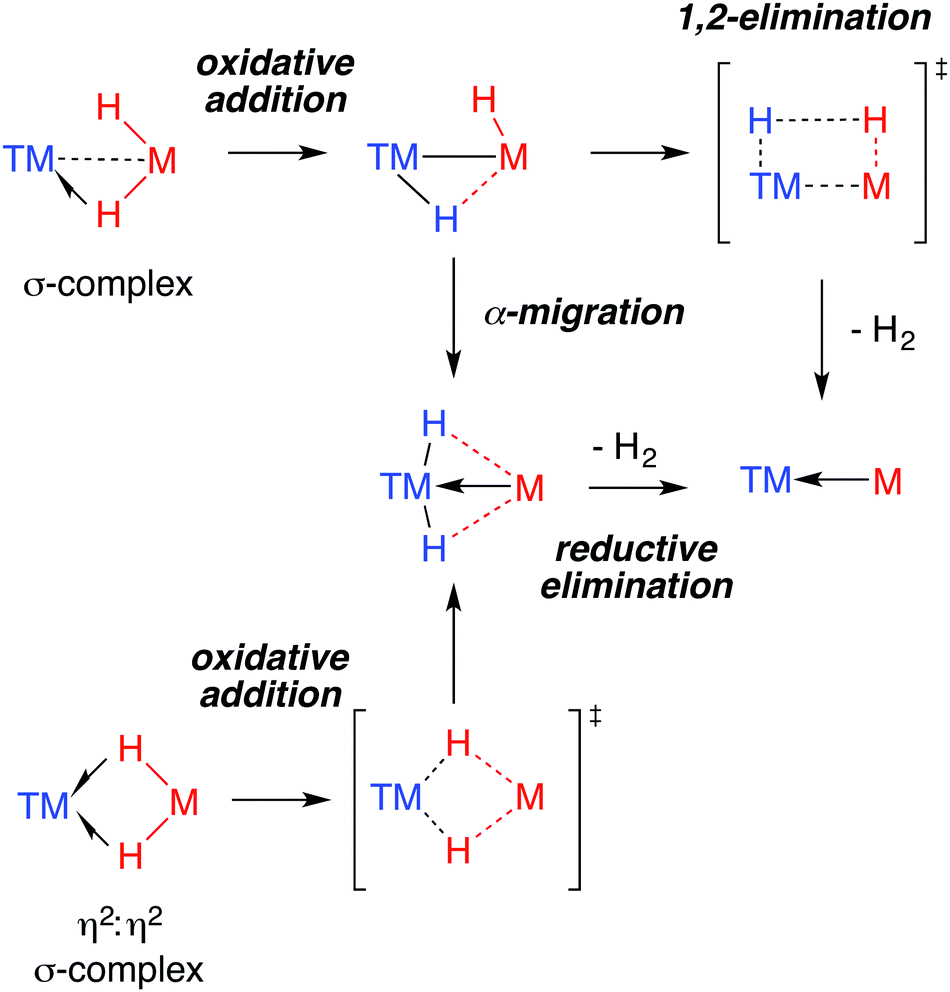 | ||
| Fig. 23 Proposed mechanisms for H2 elimination from heterobimetallic hydrides. | ||
The microscopic reverse of the 1,2-elimination of dihydrogen from a TM(H)–M(H) unit is H2 addition across a dative TM←M bond. A handful of examples of this type of reactivity are known, albeit from adducts in which the roles of the metals in the Lewis acid–base adduct are reversed. For example, the addition of dihydrogen across Pt→Al and Ru→Zn bonds to form heterobimetallic complexes is known.82,175 It remains likely that similar reactivity will be discovered for TM←M adducts. This may well be foreshadowed by the existing observations of both inter- and intramolecular addition of carbon–hydrogen bonds across in situ generated TM←M (16) and TM–M (20, 31, 44) complexes.
By considering the properties of the TM–H–Zn complexes on the reaction trajectory presented in Fig. 18, DFT studies have shown that as the hydride is transferred from zinc it becomes less hydridic and more acidic.37 While the data would suggest that a rich acid/base chemistry may be possible with heterobimetallic hydride complexes, the reactivity of these species remains understudied. The same with insertion of unsaturated substrates (e.g. alkenes, alkynes, CO, CO2, carbonyls, etc.) into 3-centre 2-electron TM–H–M bonds: it remains unclear how this chemistry will compare to the well-studied reactions of transition metal hydrides.
(iii) Potential in catalysis
Transition metal hydride complexes are known intermediates in the hydrogenation of unsaturated hydrocarbons, CO (Fischer–Tropsch), and CO2 along with numerous other small molecules.185–187 They also play a key role in hydrogenase enzymes and related catalysts for H+ reduction to H2 and have been invoked as (off-cycle) intermediates during a number of important polymerisation reactions including Ziegler–Natta catalysis.85,109To date however, defined catalytic reactions believed to involve heterobimetallic hydride complexes are limited. Systems that are catalytic in both the transition and main group metal are the most attractive. Lu and co-workers have reported the heterobimetallic complexes 107 as catalysts for alkene hydrogenation and isomerisation and demonstrated that the nature of the main group metal, M, effects the activity (Fig. 24).188 Dihydrogen activation by addition across a Ni→M bond has been proposed as a key step in hydrogenation catalysis.
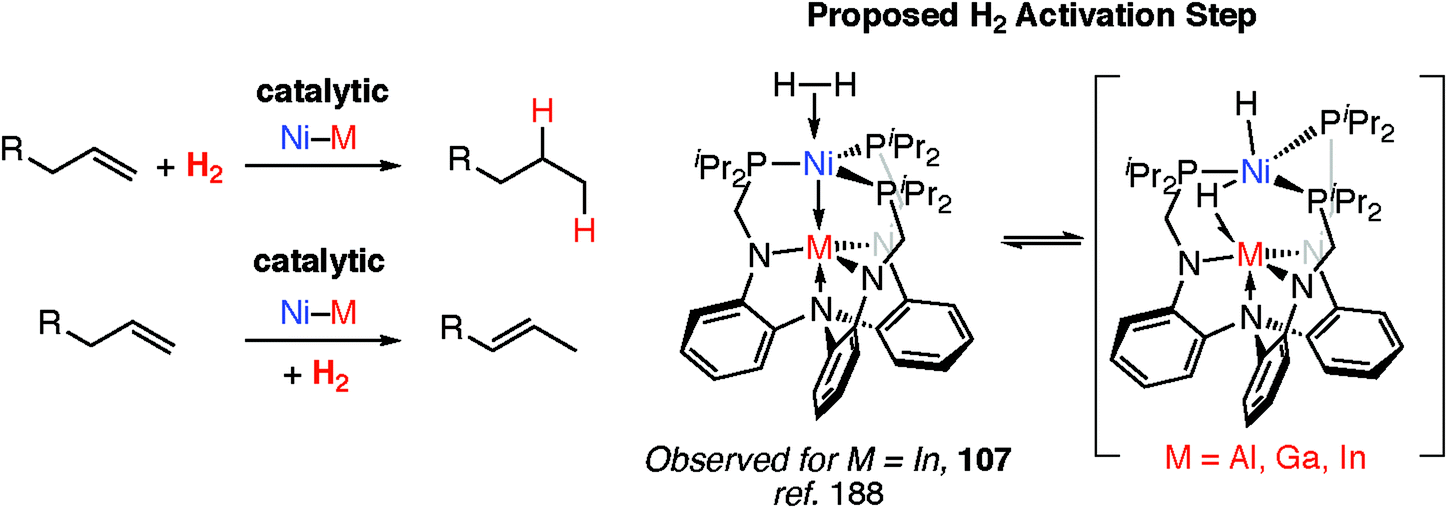 | ||
| Fig. 24 Catalytic alkene hydrogenation and isomerisation by heterobimetallic complexes. | ||
The importance of transition metal hydrides to numerous aspects of catalysis is not up for debate. Whether or not heterobimetallic complexes will be able to offer advantages over existing monometallic systems remains an open question for the community. It is clear that substantial developments in the stoichiometric and catalytic reactivity of heterobimetallic hydrides need to be made. Nevertheless, the co-location of two metals by direct metal–metal bonds and/or bridging ligands offers two opportunities in catalysis: new fundamental reactivity, and fine tuning of selection events in known reactions.
The task for the current generation of chemists is to transcribe the known methods for preparation, and the structural understanding of, heterobimetallic hydride complexes into new reactivity. While progress in this area seems to have been made in stops and starts, given the renewed interest in heterobimetallics and main group complexes for catalysis, the challenge appears timely.189,190
Acknowledgements
We are grateful to the Royal Society (MRC), EPSRC (EP/L011514/1) and ERC for funding. We are grateful to Olga Ekkert and Samantha Lau for informative discussions.Notes and references
- R. H. Crabtree, Angew. Chem., Int. Ed., 1993, 32, 789–805 CrossRef.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- G. J. Kubas, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6901–6907 CrossRef CAS PubMed.
- G. J. Kubas, J. Organomet. Chem., 2001, 635, 37–68 CrossRef CAS.
- G. J. Kubas, Acc. Chem. Res., 1988, 21, 120–128 CrossRef.
- S. Sabo-Etienne and B. Chaudret, Coord. Chem. Rev., 1998, 178-180, 381–407 CrossRef CAS.
- G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640–1649 CrossRef CAS PubMed.
- J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed.
- S. Lachaize and S. Sabo-Etienne, Eur. J. Inorg. Chem., 2006, 2115–2127 CrossRef CAS.
- G. I. Nikonov, J. Organomet. Chem., 2001, 635, 24–36 CrossRef CAS.
- J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11481–11503 RSC.
- J. Y. Corey, Chem. Rev., 2011, 111, 863–1071 CrossRef CAS PubMed.
- S. B. Duckett and R. N. Perutz, Organometallics, 1992, 11, 90–98 CrossRef CAS.
- S. Sakaki, N. Mizoe and M. Sugimoto, Organometallics, 1998, 17, 2510–2523 CrossRef CAS.
- G. J. Kubas, Catal. Lett., 2005, 104, 79–101 CrossRef CAS.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205 CrossRef CAS PubMed.
- C. E. Webster, Y. Fan, M. B. Hall, D. Kunz and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 858–859 CrossRef CAS PubMed.
- J. F. Hartwig, K. S. Cook, M. Hapke, C. D. Incarvito, Y. Fan, C. E. Webster and M. B. Hall, J. Am. Chem. Soc., 2005, 127, 2538–2552 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- A. R. Barron and G. Wilkinson, Polyhedron, 1986, 5, 1897–1915 CrossRef CAS.
- G. L. Soloveichik and B. M. Bulychev, Russ. Chem. Rev., 1983, 51, 43–60 CrossRef.
- G. Parkin, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 136, pp. 113–145 Search PubMed.
- R. A. Fischer and J. Weiß, Angew. Chem., Int. Ed., 1999, 38, 2830–2850 CrossRef PubMed.
- C. Gemel, T. Steinke, M. Cokoja, A. Kempter and R. A. Fischer, Eur. J. Inorg. Chem., 2004, 4161–4176 CrossRef CAS.
- T. Bollermann, C. Gemel and R. A. Fischer, Coord. Chem. Rev., 2012, 256, 537–555 CrossRef CAS.
- S. Gonzalez-Gallardo, G. Prabusankar, T. Cadenbach, C. Gemel, M. von Hopffgarten, G. Frenking and R. A. Fischer, Metal–Metal Bonding, 2010, pp. 147–188 Search PubMed.
- A. E. Nako, Q. W. Tan, A. J. P. White and M. R. Crimmin, Organometallics, 2014, 33, 2685–2688 CrossRef CAS.
- S. Schlecht and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9435–9443 CrossRef CAS.
- T. Kakizawa, Y. Kawano and M. Shimoi, Organometallics, 2001, 20, 3211–3213 CrossRef CAS.
- C. Ganesamoorthy, J. Weßing, C. Kroll, R. W. Seidel, C. Gemel and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7943–7947 CrossRef CAS PubMed.
- I. M. Riddlestone, S. Edmonds, P. A. Kaufman, J. Urbano, J. I. Bates, M. J. Kelly, A. L. Thompson, R. Taylor and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 2551–2554 CrossRef CAS PubMed.
- I. M. Riddlestone, J. Urbano, N. Phillips, M. J. Kelly, D. Vidovic, J. I. Bates, R. Taylor and S. Aldridge, Dalton Trans., 2013, 42, 249–258 RSC.
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin, N. Phillips, J. I. Bates and S. Aldridge, Chem. Commun., 2013, 49, 17624–17634 RSC.
- J. A. B. Abdalla, I. M. Riddlestone, J. Turner, P. A. Kaufman, R. Tirfoin, N. Phillips and S. Aldridge, Chem. – Eur. J., 2014, 20, 17624–17634 CrossRef CAS PubMed.
- I. M. Riddlestone, J. A. B. Abdalla and S. Aldridge, Advances in Organometallic Chemistry, 2015, vol. 63, pp. 1–38 Search PubMed.
- J. Turner, J. A. B. Abdalla, J. I. Bates, R. Tirfoin, M. J. Kelly, N. Phillips and S. Aldridge, Chem. Sci., 2013, 4, 4245–4250 RSC.
- O. Ekkert, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2016, 55, 16031–16034 CrossRef CAS PubMed.
- K. Ueno, T. Yamaguchi, K. Uchiyama and H. Ogino, Organometallics, 2002, 21, 2347–2349 CrossRef CAS.
- T. Muraoka and K. Ueno, Coord. Chem. Rev., 2010, 254, 1348–1355 CrossRef CAS.
- K.-R. Porschke, W. Kleimann, Y.-H. Tsay, C. Kruger and G. Wilke, Chem. Ber., 1990, 123, 1267–1273 CrossRef.
- T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299–2302 CrossRef CAS PubMed.
- T. Steinke, M. Cokoja, C. Gemel, A. Kempter, A. Krapp, G. Frenking, U. Zenneck and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 2943–2946 CrossRef CAS PubMed.
- O. Ekkert, A. J. P. White, H. Toms and M. R. Crimmin, Chem. Sci., 2015, 6, 5617–5622 RSC.
- L. J. Hounjet, D. Adhikari, M. Pink, P. J. Carroll and D. J. Mindiola, Z. Anorg. Allg. Chem., 2015, 641, 45–48 CrossRef CAS.
- L. Fohlmeister, S. Liu, C. Schulten, B. Moubaraki, A. Stasch, J. D. Cashion, L. Gagliardi and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8294–8298 CrossRef CAS PubMed.
- A. Kempter, C. Gemel and R. A. Fischer, Chem. – Eur. J., 2007, 13, 2990–3000 CrossRef CAS PubMed.
- A. Doddi, G. Prabusankar, C. Gemel, M. Winter and R. A. Fischer, Eur. J. Inorg. Chem., 2013, 3609–3615 CrossRef CAS.
- T. Cadenbach, T. Bollermann, C. Gemel and R. A. Fischer, Dalton Trans., 2009, 322–329 RSC.
- J. J. Schneider, C. Kruger, M. Nolte, I. Abraham, T. S. Ertel and H. Bertagnoli, Angew. Chem., Int. Ed., 1994, 33, 2435–2437 CrossRef.
- M. D. Fryzuk, D. H. McConville and S. J. Rettig, Organometallics, 1990, 9, 1359–1360 CrossRef CAS.
- M. D. Fryzuk, D. H. McConville and S. J. Rettig, Organometallics, 1993, 12, 2152–2161 CrossRef CAS.
- T. Cadenbach, C. Gemel, R. Schmid, M. Halbherr, K. Yusenko, M. Cokoja and R. A. Fischer, Angew. Chem., Int. Ed., 2009, 48, 3872–3876 CrossRef CAS PubMed.
- T. Cadenbach, T. Bollermann, C. Gemel, M. Tombul, I. Fernandez, M. V. Hopffgarten, G. Frenking and R. A. Fischer, J. Am. Chem. Soc., 2009, 131, 16063–16077 CrossRef CAS PubMed.
- T. Cadenbach, C. Gemel, T. Bollermann, I. Fernandez, G. Frenking and R. A. Fischer, Chem. – Eur. J., 2008, 14, 10789–10796 CrossRef CAS PubMed.
- A. Kempter, C. Gemel, T. Cadenbach and R. A. Fischer, Organometallics, 2007, 26, 4257–4264 CrossRef CAS.
- M. Molon, C. Gemel and R. A. Fischer, J. Organomet. Chem., 2014, 751, 573–578 CrossRef CAS.
- B. Buchin, C. Gemel, A. Kempter, T. Cadenbach and R. A. Fischer, Inorg. Chim. Acta, 2006, 359, 4833–4839 CrossRef CAS.
- G. Tan, T. Szilvási, S. Inoue, B. Blom and M. Driess, J. Am. Chem. Soc., 2014, 136, 9732–9742 CrossRef CAS PubMed.
- R. A. Fischer, T. Priermeier and W. Scherer, J. Organomet. Chem., 1993, 459, 65–71 CrossRef CAS.
- R. A. Fischer, M. M. Schulte, J. Weiß, L. Zsolnai, A. Jacobi, G. Huttner, G. Frenking, C. Boehme and S. F. Vyboishchikov, J. Am. Chem. Soc., 1998, 120, 1237–1248 CrossRef CAS.
- R. A. Fischer and T. Priermeier, Organometallics, 1994, 13, 4306–4314 CrossRef CAS.
- J. T. Golden, T. H. Peterson, P. L. Holland, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1998, 120, 223–224 CrossRef CAS.
- H. Brunner, P. C. Wailes and H. D. Kaesz, Inorg. Nucl. Chem. Lett., 1965, 1, 125–129 CrossRef CAS.
- A. Storr and B. S. Thomas, Can. J. Chem., 1971, 49, 2504–2507 CrossRef CAS.
- J. W. Bruno, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 444–445 CrossRef CAS.
- M. Amati and F. Lelj, Can. J. Chem., 2009, 87, 1406–1414 CrossRef CAS.
- P. C. Wailes, H. Weigold and A. P. Bell, J. Organomet. Chem., 1972, 43, C29–C31 CrossRef CAS.
- M. L. H. Green, R. E. MacKenzie and J. S. Poland, J. Chem. Soc., Dalton Trans., 1976, 1993–1994 RSC.
- M. Stender, H. Oesen, S. Blaurock and E. Hey Hawkins, Z. Anorg. Allg. Chem., 2001, 980–984 CrossRef.
- R. A. Forder and K. Prout, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1974, B30, 2312–2317 CrossRef.
- S. J. Rettig, A. Storr, B. S. Thomas and J. Trotter, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1974, B30, 666–672 CrossRef.
- D. E. Crotty, T. J. Anderson, M. D. Glick and J. P. Oliver, Inorg. Chem., 1977, 16, 2346–2350 CrossRef CAS.
- R. L. Geerts, J. C. Huffman and K. G. Caulton, Inorg. Chem., 1986, 25, 590–591 CrossRef CAS.
- M. A. Porai-Koshits, A. S. Antsyshkina, A. A. Pasynskii, G. G. Sadikov, Y. V. Skripkin and V. N. Ostrikova, Inorg. Chim. Acta, 1979, 34, L285–L287 CrossRef CAS.
- F. N. Tebbe, J. Am. Chem. Soc., 1973, 95, 5412–5414 CrossRef CAS.
- P. H. M. Budzelaar, A. A. H. Van Der Zeijden, J. Boersma, G. J. M. Van Der Kerk, A. L. Spek and A. J. M. Duisenberg, Organometallics, 1984, 3, 159–163 CrossRef CAS.
- P. H. M. Budzelaar, K. H. Den Haan, J. Boersma, G. J. M. Van Der Kerk and A. L. Spek, Organometallics, 1984, 3, 156–159 CrossRef CAS.
- M. Ohashi, K. Matsubara, T. Iizuka and H. Suzuki, Angew. Chem., Int. Ed., 2003, 49, 937–940 CrossRef PubMed.
- M. Ohashi, K. Matsubara and H. Suzuki, Organometallics, 2007, 26, 2330–2339 CrossRef CAS.
- K. Prout and R. A. Forder, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1975, B31, 852–856 CrossRef CAS.
- M. L. H. Green, G. A. Moser, I. Packer, F. Petit, R. A. Forder and K. Prout, J. Chem. Soc., Chem. Commun., 1974, 839–840 RSC.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed.
- R. A. Fischer, D. Weiß, M. Winter, I. Müller, H. D. Kaesz, N. Fröhlich and G. Frenking, J. Organomet. Chem., 2004, 689, 4611–4623 CrossRef CAS.
- B. E. Cowie, F. A. Tsao and D. J. H. Emslie, Angew. Chem., Int. Ed., 2014, 54, 2165–2169 CrossRef PubMed.
- L. V. Parfenova, P. V. Kovyazin, I. E. Nifant’ev, L. M. Khalilov and U. M. Dzhemilev, Organometallics, 2015, 34, 3559–3570 CrossRef CAS.
- V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2006, 25, 519–522 CrossRef CAS.
- J. Kopf, H. J. Vollmer and W. Kaminsky, Cryst. Struct. Commun., 1980, 9, 985 CAS.
- A. I. Sizov, T. M. Zvukova, A. A. Gorkovskii, Z. A. Starikova and B. M. Bulychev, Russ. Chem. Bull., 2004, 53, 2175–2178 CrossRef CAS.
- T. M. Arkhireeva, V. K. Bel'skii, B. M. Bulychev and G. L. Soloveichik, Russ. Chem. Bull., 1986, 35, 2585–2587 CrossRef.
- D. Schädle, C. Schädle, D. Schneider, C. Maichle-Mössmer and R. Anwander, Organometallics, 2015, 34, 4994–5008 CrossRef.
- C. Schädle, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Organometallics, 2015, 34, 2667–2675 CrossRef.
- C. Schädle, D. Schädle, K. Eichele and R. Anwander, Angew. Chem., Int. Ed., 2013, 52, 13238–13242 CrossRef PubMed.
- M. G. Klimpel, P. Sirsch, W. Scherer and R. Anwander, Angew. Chem., Int. Ed., 2003, 42, 574–577 CrossRef CAS PubMed.
- S. Y. Knjazhanski, B. M. Bulychev, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1987, 327, 173–179 CrossRef.
- C. Schädle, A. Fischbach, E. Herdtweck, K. W. Törnroos and R. Anwander, Chem. – Eur. J., 2013, 19, 16334–16341 CrossRef PubMed.
- C. Schädle and R. Anwander, Eur. J. Inorg. Chem., 2013, 3302–3306 CrossRef.
- E. B. Lobkovskii, G. L. Soloveichik, A. B. Erofeev, B. M. Bulychev and V. K. Bel'skii, J. Organomet. Chem., 1982, 235, 151–159 CrossRef CAS.
- E. Lobkovsky, G. L. Soloveychik, B. M. Bulychev, A. B. Erofeev, A. I. Gusev and N. I. Kirillova, J. Organomet. Chem., 1983, 254, 167–172 CrossRef.
- V. K. Bel'skii, B. M. Bulychev, A. B. Erofeev and G. L. Soloveichik, J. Organomet. Chem., 1984, 268, 107–111 CrossRef.
- V. K. Belsky, A. B. Erofeev, B. M. Bulychev and G. L. Soloveichik, J. Organomet. Chem., 1984, 265, 123–133 CrossRef.
- S. Y. Knjazhanski, B. M. Bulychev, O. K. Kireeva, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1991, 414, 11–22 CrossRef.
- V. K. Belsky, Y. K. Gun’ko, B. M. Bulychev and G. L. Soloveichik, J. Organomet. Chem., 1991, 419, 299–310 CrossRef.
- Y. K. Gun’ko, B. M. Bulychev, A. I. Sizov, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1990, 390, 153–158 CrossRef.
- A. B. Erofeev, B. M. Bulychev, V. K. Bel'skii and G. L. Soloveichik, J. Organomet. Chem., 1987, 335, 189–199 CrossRef CAS.
- S. Y. Knjazhanski, E. B. Lobkovsky, B. M. Bulychev, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1991, 419, 311–324 CrossRef.
- M. F. Lappert, A. Singh, J. L. Atwood and W. E. Hunter, J. Chem. Soc., Chem. Commun., 1983, 206–207 RSC.
- K. D. Conroy, W. E. Piers and M. Parvez, Organometallics, 2009, 28, 6228–6233 CrossRef CAS.
- Y. K. Gun'ko, B. M. Bulychev, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1992, 440, 47–52 CrossRef.
- H. G. Alt and A. Köppl, Chem. Rev., 2000, 100, 1205–1222 CrossRef CAS PubMed.
- L. I. Shoer, K. I. Gell and J. Schwartz, J. Organomet. Chem., 1977, 136, C19–C22 CrossRef CAS.
- S. M. Baldwin, J. E. Bercaw and H. H. Brintzinger, J. Am. Chem. Soc., 2008, 130, 17423–17433 CrossRef CAS PubMed.
- U. M. Dzhemilev and A. G. Ibragimov, Russ. Chem. Rev., 2012, 81, 524–548 CrossRef.
- K. P. Bryliakov, E. P. Talsi, A. Z. Voskoboynikov, S. J. Lancaster and M. Bochmann, Organometallics, 2008, 27, 6333–6342 CrossRef CAS.
- S. M. Baldwin, J. E. Bercaw, L. M. Henling, M. W. Day and H. H. Brintzinger, J. Am. Chem. Soc., 2011, 133, 1805–1813 CrossRef CAS PubMed.
- S. M. Baldwin, J. E. Bercaw and H. H. Brintzinger, J. Am. Chem. Soc., 2010, 132, 13969–13971 CrossRef CAS PubMed.
- K. Khan, C. L. Raston, J. E. McGrady, B. W. Skelton and A. H. White, Organometallics, 1997, 16, 3253–3254 CrossRef.
- R. J. Wehmschulte and P. P. Power, Polyhedron, 1999, 18, 1885–1888 CrossRef CAS.
- N. Etkin, A. J. Hoskin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 11420–11424 CrossRef CAS.
- S. Yow, S. J. Gates, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2012, 51, 12559–12563 CrossRef CAS PubMed.
- M. J. Butler, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2016, 55, 6951–6953 CrossRef CAS PubMed.
- N. Etkin and D. W. Stephan, Organometallics, 1998, 17, 763–765 CrossRef CAS.
- E. B. Lobkovskii, G. L. Soloveichik, B. M. Bulychev, R. G. Gerr and Y. T. Struchkov, J. Organomet. Chem., 1984, 270, 45–51 CrossRef CAS.
- E. B. Lobkovskii, G. L. Soloveichik, A. J. Sisov, B. M. Bulychev, A. I. Gusev and N. I. Kirillova, J. Organomet. Chem., 1984, 265, 167–173 CrossRef CAS.
- V. K. Bel'sky, A. I. Sizov, B. M. Bulychev and G. L. Soloveichik, J. Organomet. Chem., 1985, 280, 67–80 CrossRef.
- T. Y. Sokolova, A. I. Sizov, B. M. Bulychev, E. A. Rozova, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1990, 288, 11–19 CrossRef.
- A. I. Sizov, T. M. Zvukova, V. K. Belsky and B. M. Bulychev, Russ. Chem. Bull., 1998, 47, 1186–1192 CrossRef CAS.
- A. I. Sizov, T. M. Zvukova, B. M. Bulychev and V. K. Belsky, J. Organomet. Chem., 2000, 603, 167–173 CrossRef CAS.
- R. Sun, J. Liu, S. Yang, M. Chen, N. Sun, H. Chen, X. Xie, X. You, S. Li and Y. Liu, Chem. Commun., 2015, 51, 6426–6429 RSC.
- J. Thomas, M. Klahn, A. Spannenberg and T. Beweries, Dalton Trans., 2013, 42, 14668–14672 RSC.
- A. I. Sizov, I. V. Molodnitskaya, B. M. Bulychev, V. K. Bel'skii and G. L. Soloveichik, J. Organomet. Chem., 1988, 344, 185–193 CrossRef CAS.
- A. I. Sizov, T. M. Zvukova, V. K. Belsky and B. M. Bulychev, J. Organomet. Chem., 2001, 619, 36–42 CrossRef CAS.
- A. I. Sizov, T. M. Zvukova, V. K. Belsky and B. M. Bulychev, Russ. Chem. Bull., 2001, 50, 2200–2202 CrossRef CAS.
- L. J. Guggenberger and F. N. Tebbe, J. Am. Chem. Soc., 1973, 95, 7870–7872 CrossRef CAS.
- F. N. Tebbe and L. J. Guggenberger, J. Chem. Soc., Chem. Commun., 1973, 227–228 RSC.
- S. I. Troyanov, V. Varga and K. Mach, J. Organomet. Chem., 1993, 461, 85–90 CrossRef CAS.
- R. Gyepes, K. Mach, I. Cisarova, J. Loub, J. Hiller and P. Sindelar, J. Organomet. Chem., 1995, 497, 33–41 CrossRef CAS.
- M. Horacek, J. Hiller, U. Thewalt, M. Polášek and K. Mach, Organometallics, 1997, 16, 4185–4191 CrossRef CAS.
- V. Kupfer, U. Thewalt, M. Horacek, L. Petrusova and K. Mach, Inorg. Chem. Commun., 1999, 2, 540–544 CrossRef CAS.
- M. Horáček, I. Cisarova, J. Cejka, J. Karban, L. Petrusova and K. Mach, J. Organomet. Chem., 1999, 577, 103–112 CrossRef.
- M. Horáček, P. Štěpnička, J. Kubišta, K. Fejfarová, R. Gyepes and K. Mach, Organometallics, 2003, 22, 861–869 CrossRef.
- K. Mach, R. Gyepes, M. Horáček, L. Petrusová and J. Kubišta, Collect. Czech. Chem. Commun., 2003, 68, 1877–1896 CrossRef CAS.
- Q. F. Mokuolu, P. A. Duckmanton, A. J. Blake, C. Wilson and J. B. Love, Organometallics, 2003, 22, 4387–4389 CrossRef CAS.
- E. B. Lobkovskii, G. L. Soloveichik, A. I. Sizov and B. M. Bulychev, J. Organomet. Chem., 1985, 280, 53–66 CrossRef CAS.
- E. B. Lobkovskii, A. I. Sizov, B. M. Bulychev and I. V. Sokolova, J. Organomet. Chem., 1987, 319, 69–75 CrossRef CAS.
- H. Lee, T. Hascall, P. J. Desrosiers and G. Parkin, J. Am. Chem. Soc., 1998, 120, 5830–5831 CrossRef CAS.
- G. Erker, M. Albrecht, C. Kruger, S. Werner, P. Binger and F. Langhauser, Organometallics, 1992, 11, 3517–3525 CrossRef CAS.
- R. Gleiter, I. Hyla-Kryspin, S. Niu and G. Erker, Angew. Chem., Int. Ed., 1993, 32, 754–756 CrossRef.
- M. Albrecht, G. Erker and C. Kruger, Synlett, 1993, 441–448 CrossRef CAS.
- G. Bender, T. Wiegand, H. Eckert, R. Fröhlich, C. G. Daniliuc, C. Mück-Lichtenfeld, S. Ndambuki, T. Ziegler, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2012, 51, 8846–8849 CrossRef CAS PubMed.
- P. Arndt, A. Spannenberg, W. Baumann, S. Becke and U. Rosenthal, Eur. J. Inorg. Chem., 2001, 2885–2890 CrossRef CAS.
- V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2004, 23, 4160–4165 CrossRef CAS.
- V. V. Burlakov, K. Kaleta, T. Beweries, P. Arndt, W. Baumann, A. Spannenberg, V. B. Shur and U. Rosenthal, Organometallics, 2011, 30, 1157–1161 CrossRef CAS.
- P. Arndt, A. Spannenberg, W. Baumann, V. V. Burlakov, U. Rosenthal, S. Becke and T. Weiss, Organometallics, 2004, 23, 4792–4795 CrossRef CAS.
- R. M. Waymouth, B. D. Santarsiero, R. J. Coots, M. J. Bronikowski and R. H. Grubbs, J. Am. Chem. Soc., 1986, 108, 1427–1441 CrossRef CAS.
- G. S. Girolami, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Am. Chem. Soc., 1983, 105, 6752–6753 CrossRef CAS.
- G. S. Girolami, C. G. Howard, G. Wilkinson, H. M. Dawes, M. Thornton-Pett, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1985, 921–929 RSC.
- A. R. Barron, G. Wilkinson, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1987, 837–846 RSC.
- R. Gilbert-Wilson, L. D. Field and M. Bhadbhade, Inorg. Chem., 2014, 53, 12469–12479 CrossRef CAS PubMed.
- G. S. Girolami, Organometallics, 1997, 2987–2994 Search PubMed.
- M. Plois, T. Wiegand and R. Wolf, Organometallics, 2012, 31, 8469–8477 CrossRef CAS.
- M. Plois, R. Wolf, W. Hujo and S. Grimme, Eur. J. Inorg. Chem., 2013, 3039–3048 CrossRef CAS.
- T. N. Gieshoff, M. Villa, A. Welther, M. Plois, U. Chakraborty, R. Wolf and A. Jacobi von Wangelin, Green Chem., 2015, 17, 1408–1413 RSC.
- A. R. Barron, M. B. Hursthouse, M. Motevalli and G. Wilkinson, J. Chem. Soc., Chem. Commun., 1985, 664–665 RSC.
- A. R. Barron and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1986, 279–285 RSC.
- A. R. Barron, D. Lyons, G. Wilkinson, M. Motevalli, A. J. Howes and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1986, 279–285 RSC.
- D. H. Lee, H. Suzuki and Y. Moro-oka, J. Organomet. Chem., 1987, 330, C20–C22 CrossRef CAS.
- M. D. Fryzuk, G. K. B. Clentsmith and S. J. Rettig, Inorg. Chim. Acta, 1997, 259, 51–59 CrossRef CAS.
- V. Saboonchian, G. Wilkinson, B. Hussain-Bates and M. B. Hursthouse, Polyhedron, 1991, 10, 595–603 CrossRef CAS.
- G. I. Nikonov, L. G. Kuzmina and J. A. K. Howard, J. Chem. Soc., Dalton Trans., 2002, 3037–3046 RSC.
- T. J. McNeese, S. S. Wreford and B. M. Foxman, J. Chem. Soc., Chem. Commun., 1978, 500–502 RSC.
- O. Ekkert, S. D. A. Strudley, A. Rozenfeld, A. J. P. White and M. R. Crimmin, Organometallics, 2014, 33, 7027–7030 CrossRef CAS.
- W. A. Skupinski, J. C. Huffman, J. W. Bruno and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 8128–8136 CrossRef CAS.
- K. Knox and A. P. Ginsberg, Inorg. Chem., 1964, 3, 555–558 CrossRef CAS.
- M. Oishi, T. Endo, M. Oshima and H. Suzuki, Inorg. Chem., 2014, 53, 5100–5108 CrossRef CAS PubMed.
- I. M. Riddlestone, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2016, 138, 11081–11084 CrossRef CAS PubMed.
- M. Molon, C. Gemel and R. A. Fischer, Eur. J. Inorg. Chem., 2013, 3616–3622 CrossRef CAS.
- V. Riera, M. A. Ruiz, A. Tiripicchio and M. Tiripicchio-Camellini, Organometallics, 1993, 12, 2962–2972 CrossRef CAS.
- R. L. Bansemer, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1983, 105, 6163–6164 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- P. Klufers and U. Wilhem, J. Organomet. Chem., 1991, 421, 39–54 CrossRef.
- M. Hamdaoui, M. Ney, V. Sarda, L. Karmazin, C. Bailly, N. Sieffert, S. Dohm, A. Hansen, S. Grimme and J.-P. Djukic, Organometallics, 2016, 35, 2207–2223 CrossRef CAS.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS PubMed.
- I. Atheaux, B. Donnadieu, V. Rodriguez, S. Sabo-Etienne, B. Chaudret, K. Hussein and J.-C. Barthelat, J. Am. Chem. Soc., 2000, 122, 5664–5665 CrossRef CAS.
- M. C. Lipke and T. D. Tilley, Angew. Chem., Int. Ed., 2012, 51, 11115–11121 CrossRef CAS PubMed.
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS PubMed.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 16, 8873–8911 CrossRef PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- N. P. Mankad, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed.
- R. J. Eisenhart, L. J. Clouston and C. C. Lu, Acc. Chem. Res., 2015, 48, 2885–2894 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc05702k |
| This journal is © The Royal Society of Chemistry 2017 |