
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopeptide scaffolds in carbohydrate-based synthetic vaccines
Carlo
Pifferi
a,
Nathalie
Berthet
a and
Olivier
Renaudet
 *ab
*ab
aUniv. Grenoble Alpes, CNRS, DCM UMR 5250, F-38000 Grenoble, France. E-mail: olivier.renaudet@univ-grenoble-alpes.fr
bInstitut Universitaire de France, 103 boulevard Saint-Michel, 75005 Paris, France
First published on 9th March 2017
Abstract
Cyclopeptides have been recently used successfully as carriers for the multivalent presentation of carbohydrate and peptide antigens in immunotherapy. Beside their synthetic versatility, these scaffolds are indeed interesting due to their stability against enzyme degradation and low immunogenicity. This mini-review highlights the recent advances in the utilization of cyclopeptides to prepare fully synthetic vaccines prototypes against cancers and pathogens.
Introduction
Carbohydrates represent the most abundant and complex class of biomolecules in nature. For decades, our knowledge in this field has been limited to their metabolic or structural function, but their scientific interest has been reassessed more recently in light of their primary relevance in cell–cell communication, host–pathogen interaction, cancer metastasis or fertilization. Carbohydrates are usually expressed on the cellular surface conjugated to membrane protein or lipid, composing the glycocalyx (Fig. 1) which provides a characteristic “glyco-signature” for both mammalian cells and pathogens1 and participates to diverse biological events. For example, they are involved in the initial infection step of pathogens by adhering to proteins expressed at the surface of host cells. Moreover infected organisms detect the presence of pathogens through recognition of carbohydrates and pattern recognition receptors (PRRs) which stimulates defense responses by the host.2 Carbohydrate–protein interactions play therefore key roles both for pathogen to initiate infection and for the host to trigger immune response. By contrast, the majority of human cancers are characterized by aberrant glycosylation, i.e. tumor cells may over-express truncated versions of oligosaccharides, unusual terminal oligosaccharide sequences and show an increased sialylation of cell-surface glycolipids and O- and N-linked glycoproteins. The expression of these altered glycans, also called tumor-associated carbohydrate antigens (TACAs) participates in various stages of the tumour progression and is associated with malignancy. For these reasons, these oligosaccharides represent relevant structures for the design of synthetic vaccines. It should be mentioned that the chemical synthesis of carbohydrate antigens is a very large topic and will not be covered in this manuscript. We suggest the readers to refer to excellent reviews in this field.3–6 | ||
| Fig. 1 Examples for (a) bacterial antigen repeating units and (b) tumor associated carbohydrate antigens. | ||
Problems associated with carbohydrate-based vaccines
The major hurdle associated with the development of carbohydrate-based vaccines is however due to the poor quality of antibody responses to carbohydrates.7 The principal cause of this problem is the incapacity of carbohydrate antigens to stimulate T cell dependent response.8 Indeed they are able to activate B cells upon binding to membrane receptors (BCR) such as immunoglobulins, which induces their clustering and leads to the B cell activation (Fig. 2a). Except in rare cases,9 this primary response results in the production of low affinity and short-living IgM whereas no memory effect is induced due to the absence of T cells stimulation. | ||
| Fig. 2 (a) Carbohydrate binding to B-cell receptors, initiating a weak T-cell independent immunological response; (b) activation of Th cells through antigen presenting cells (APCs: dendritic cells, macrophages and B-cells); (c) T-cell dependent immune response. | ||
To achieve antibody class switching, B cells need the participation of helper T cells (Th cells). This requires the processing and presentation of peptidic antigens (CD4+ epitopes) by MHC-II molecules by antigen presenting cells (APCs) such as B cells, followed by its recognition by the surface protein CD4+ expressed by Th cells.
Design of synthetic vaccines
Semi-synthetic approach
As mentioned above, T cells-dependent responses are typically generated by protein and peptide epitopes. For this reason, carbohydrate-based vaccine are classically prepared by coupling glycans to immunogenic carrier proteins,5,10–14 such as keyhole limpet hemocyanin (KLH), ovalbumin (OVA), bovine serum albumin (BSA) or tetanus toxoid (TT). These proteins indeed provide CD4+ epitopes upon processing by APCs whose combination with carbohydrate epitope is now known to trigger a long-lasting and IgG-mediated antibody response that could not be achieved by the separated administration of these single elements.2,15–17 The efficiency of the immune response induced by these carbohydrate–protein conjugates depends on several factors which all contribute to the structure of the vaccine: the size and the total number (density) of conjugated carbohydrate epitopes, the nature of the carrier protein, the type and the length of the linker between the carbohydrate and the carrier.Many semi-synthetic vaccines have been developed on these structural bases. For example, Kunz's and Li's groups have synthesized vaccine candidates composed of a MUC1 glycopeptide displaying both the Tn and STn antigens,18–21 which was further conjugated to the bovine serum albumin (BSA) carrier. Immunization of Balb/c mice was found to promote a remarkable Ab response with predominant IgG1 isotype antibodies and a strong binding to breast MCF-7 tumor cell line.22 However they show some major disadvantages. First, the difficulty to control the conjugation chemistry may lead to ambiguities in composition and structure and batch to batch variations of the vaccine, and as a consequence, the reproducibility of the immune response. The second major drawback is related to the use of carrier proteins that are often highly immunogenic by themselves and will necessarily elicit strong immune responses, thus leading to carrier-induced epitope suppression. The result is an increase in the antibody response to the carrier and a decrease in the response to conjugated peptides or polysaccharides.23–29 In other words, the grafted epitope represents only a small part of the total conjugate, and it is randomly distributed on the protein carrier surface, which contains several Th cell epitopes.30 Therefore, immune responses to the carrier molecule may lead to a low level of the desired antibodies as compared to the total amount of antibodies produced.
Fully synthetic approach
With the aim to circumvent these drawbacks, Leclerc and co-workers has designed fully synthetic multiple antigen glycopeptides31 (MAGs) composed of clustered Tn antigens and CD4+ peptide epitope from the type I-poliovirus protein (PV) within a well-defined lysine-based dendritic core, paving the way to the fully synthetic approach in vaccine design. This new system offers several advantages: (i) the carbohydrate content is much higher than in traditional protein conjugates; (ii) the core dendron, representing a minor fraction of the total construct, is non immunogenic thus avoiding unwanted immune responses, and (iii) the resulting construct has a precise and well-characterized chemical structure. After these key findings in 1997, more attention has been focused on subunit of vaccines, which are devoid of any unnecessary immunogenic components, combining only elements indispensable for evoking an antigen-specific immune response.A key element in this fully synthetic approach is the central scaffold which should ensure the correct presentation of the different units, without interfering with their activity and possibly improving recognition properties by means of multivalent presentation. In this mini-review we focused our attention on the various peculiarities that make cyclopeptide interesting scaffolds for carbohydrate-based vaccines candidates.
Fully synthetic vaccine based on cyclopeptide scaffolds
The group of Dumy and Renaudet have selected cyclopeptide carriers to construct synthetic vaccines.32 In particular, they focused their attention to the RAFT cyclodecapeptide platform (Regioselectively Addressable Functionalised Template)33–35 which is composed of two adjacent proline-glycine as β-turn inducers that constrain the backbone conformation into an antiparallel β sheet (Fig. 3). | ||
| Fig. 3 RAFT platform. | ||
The platform can thus be easily prepared by solid phase peptide synthesis of the linear sequence, followed by cyclization in solution, with high level of efficiency and reproducibility. Up to six lysine residues can be introduced in the decapeptide sequence, in positions 3-4-5-8-9 and 10. The resulting scaffold thus present two separate spatial domains where the Lys 3-5-8-10 are oriented in the upper plane, while the Lys 4-9 residues delineate a second domain in the opposite plane. Lysine side-chain can be orthogonally functionalized by using orthogonal protecting groups (e.g. Boc, Alloc, Dde, etc.).36 Moreover, modified Lys can be introduced as building blocks during SPPS, giving readily available orthogonal functionalities for a further extension of the platform.
Together with these structural features, the cyclic nature of such scaffolds offers an improved stability towards enzymatic degradation compared to linear analogues, which makes cyclopeptides attractive scaffolds for biological application. For example, it was recently proven suitable in diverse research fields, in particular for the synthesis of multivalent glycosylated structures with nanomolar affinity towards lectins.37–41
For all these reasons, cyclopeptides represent interesting carriers to conjugate carbohydrate and peptide epitopes for the preparation of fully synthetic carbohydrate vaccines.
Anti-cancer vaccine design
Renaudet and Dumy in collaboration with the group of Leclerc have developed a first generation of anticancer vaccine prototypes by grafting four Tn antigen analogues as B-cell epitope, and the CD4+ peptide from the type 1 poliovirus as T-cell epitope. Tn antigen is one of the most overexpressed carbohydrate antigen found in the membrane-bound glycoprotein MUC-1 (Fig. 4).42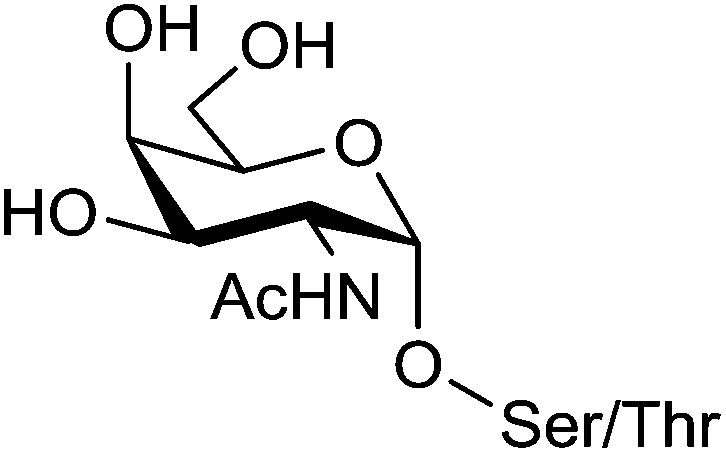 | ||
| Fig. 4 Tn antigen. | ||
The native Tn antigen is depicted as a αGalNAc unit linked to a Ser/Thr residue through a glycosidic bond. For synthetic considerations, Renaudet and Dumy have decided to introduce an oxyamine function on the anomeric carbon of the αGalNAc unit to conjugate four copies of this Tn analogue on the upper side of the RAFT scaffold by oxime ligation.43 The lower face of the construct was functionalized with the PV103–115 peptide sequence from type I-poliovirus protein (Fig. 5; compound 1), since it was previously proved efficient in triggering Th cells for the induction of antibody responses in BALB/c mice models.44 As for the conjugation of the Tn analogue, the peptide antigen was attached to the scaffold by oxime ligation. This strategy has proved efficient for the convergent assembly of diverse bioconjugates and was found compatible with a wide range of chemical functions without requiring any need of coupling agents.45–57 Moreover, the oxime bond has been shown to be stable under physiological conditions.58–60
 | ||
| Fig. 5 Structure of a clustered Tn antigen analogue-based vaccine candidate. | ||
The B cell antigenicity was assessed by ELISA using two monoclonal antibodies (mAbs) specific for the Tn antigen: an IgG3 (6E11) and an IgM (83D4). They were both able to interact with compound 1, but not with the control molecules lacking the sugar moiety or presenting another saccharide. This clearly demonstrates that the recognition is due to the unnatural Tn analogue. Its multivalent presentation at the cyclopeptide surface indeed seems able to mimic the carbohydrate part of the repeated glycosylated serine/threonine antigen unit found on natural tumor-associated mucins. Therefore, the Tn analogue represents suitable model for humoral stimulation as other reported analogues.61–64
T cell antigenicity assays indicated that the response (IL-2 production) induced by the vaccine candidate 1 was enhanced at significantly lower doses (10−4 M) compared to the response obtained with the unglycosylated construct (1 M). Indeed, T cell stimulation was obtained with about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold less dose of 1 compared to the unglycosylated control, suggesting that the presence of the Tn antigen increases the presentation of the PV peptide by MHC class II molecules. This result further confirms previous observations on the enhanced T cell stimulation induced by peptides bearing the Tn antigen.65,66
000-fold less dose of 1 compared to the unglycosylated control, suggesting that the presence of the Tn antigen increases the presentation of the PV peptide by MHC class II molecules. This result further confirms previous observations on the enhanced T cell stimulation induced by peptides bearing the Tn antigen.65,66
Immunogenicity and the ability to elicit IgG antibodies were next evaluated with the construct 1 in BALB/c mice. Sera collected from immunized mice was tested by ELISA and showed a strong recognition of the clustered Tn analogue moiety. Interestingly, only 0.1–1% of the antibodies raised by the vaccine candidate 1 recognized the construct devoid of the GalNAc moiety, thus demonstrating a low immunogenicity of the scaffold itself. To clearly determine whether the antibodies elicited by the vaccine candidate 1 are able to recognize the native form of Tn, flow cytometry has been used to assess their binding to the human Jurkat tumor cell line expressing the native Tn antigen. Sera from mice immunized with 1 were able to recognize the Jurkat cells, whereas a control compound devoid of Tn did not. These results show the ability of the cyclopeptide-based constructs bearing B cell and CD4+ T cell epitopes to elicit an immune response specifically directed against the native form of carbohydrate antigen displayed by cancer cells.
On the basis of these promising results, a second generation of vaccine prototypes has been designed by the same group. These compounds combined for the first time four essential components within a single molecule: a cluster of carbohydrate B-cell antigen, two peptides capable to stimulate CD4+ and CD8+ T-cells and an immunoadjuvant, the palmitic acid (PAM). The role of the CD8+ epitope is to activate cytotoxic T cells (CTLs) which also require CD4+ T cells for help, prime and sustain their cytotoxic functions.67–73 The palmitic acid derived from a immunologically active lipoprotein of Escherichia coli72,74 used as adjuvant75–82 is a ligand for toll like receptor 2 (TLR-2) expressed on the surface of antigen presenting cells that stimulates their maturation, cytokine production, and it is likely to promote the uptake of the conjugated vaccine prototype.
This second generation of vaccine prototype was constructed using a convergent ligation chemistry based on both oxime and disulfide bond formation, respectively for anchoring B-cell epitopes and the lipopeptidic chain to the cyclopeptide platform. All three compounds (Fig. 6; compounds 2, 3 and 4) synthesized in this series show a clustered Tn analogue B-cell epitope. In compound 2, the OVA257–264 peptide (SIINFEKL)83 was used as a target CD8+ T-cell epitope, and synthesized in line with the Pan-DR universal CD4+ Th peptide (PADRE, {dA}K{Cha}VAAWTLKAA{dA}{Ahx}).84 The resulting construct was extended at the N-terminal end with a palmitic acid moiety to confer the self-adjuvanting property of this four-component glyco-lipo-peptide vaccine.
 | ||
| Fig. 6 Second generation of prototype vaccine composed of four components. | ||
The immunological properties of the glycolipopeptide 2 were investigated in the MO5/BALB/c tumor mouse model. A strong production of antibodies that recognize human tumor cell lines (MCF7) as well as a stimulation of T-cells without administration of external adjuvant were clearly shown. More interestingly, an improved survival rate and a significant tumor regression were also observed. Among twenty mice challenged subcutaneously with MO5 cells, none of them developed a tumor after 90 days. In addition, all the mice survived after this delay, whereas the survival was less then 30% in non-immunized control group.
Compounds 3 and 4 have been further synthesized by the same group on the model 2, except for the peptide that contains a HER420–429 sequence (PDSLRDLSVFK) from the HER-2/neu glycoprotein that is over-expressed by breast carcinomas. This peptide act as a CD8+ T cell epitope, replacing the OVA protein. Moreover, the palmitoyl moiety was attached at the N-terminal end (linear glyco-lipo-peptide 3), or in between the CD4+ and CD8+ T cell epitope sequences (branched glyco-lipo-peptide 4). The uptake, processing and cross-presentation pathway of 3 and 4 were investigated to assess whether the position of the lipid moiety would affect the B- and T-cell immunogenicity and protective efficacy. While more potent HER420–429-specific IFN-γ CD8+ T cell responses was induced by the linear construct 3, the branched compound 4 generated stronger tumor-specific IgG responses. Furthermore, although both constructs enter dendritic cells via TLR2 and induce their maturation, each construct appear to be processed and presented to T cells differently. Thus, the position of the lipid moiety within synthetic glyco-lipo-peptide constructs profoundly affects the phenotypic and functional maturation process of DCs, the processing of the glyco-peptide molecules and their cross-presentation in DCs, as well as the magnitude of IgG and CD8+ T-cell responses. Although cellular and molecular mechanisms underlying the immunogenicity of 3 and 4 remain to be fully elucidated, data suggests that the position of the lipid moiety strongly impact the immunogenicity of these constructs and is superior compared to non-lipidated analogues. It indeed not only affected the uptake and cross-presentation pathways in DCs, but modulated the magnitude of antitumor antibody and CD8+ T-cell protective immunity. It can be expected that the presence of the lipid tail may promote the self-organisation of the vaccine candidate in solution, which may impact the resulting immunogenicity. However, this aspect has not been discussed by the authors.
A third generation of vaccine construct was elaborated using the same scaffold. This vaccine prototype includes four clustered Tn-antigen mimetcs as B-cell epitopes and the PADRE sequence in line with the OVA257–264 peptide, respectively as CD4+ Th and CD8+ CTL epitopes conjugated on the cyclopeptide carrier (Fig. 7; compound 5).85 It has been hypothesized that TACA-based vaccines displaying these mimetics instead of native Tn antigens could be more stable towards enzymatic degradation by endogenous glycosidases.
 | ||
| Fig. 7 Structure of the clustered α-Tn antigen mimetic based vaccine prototype. | ||
The bioactive Tn-antigen mimetic, developed by Nativi and co-workers, is a 2-deoxy-2-thio-α-O-galactoside that retains the 4C1 chair conformation of the native antigen (Fig. 8).86
 | ||
| Fig. 8 Structure of the α-Tn antigen mimetic residue. | ||
It was conjugated through amidic linkage to the Lys residues on the scaffold, and the immunostimulant peptide was linked on the lower domain of the scaffold via disulfide bond ligation.
The safety and immunogenicity were evaluated in B10.D1 mice inoculated with 5 in presence of CpG1826 adjuvant. High levels of mucin-specific IgG/IgM antibodies were induced (IgG/IgM titers >8000), and approximately half of the titer was still present in the serum 240 days after the last immunization. Moreover, antibodies were found to bind to human (MCF7) cancer cell lines expressing the native carbohydrate antigens. Immunotherapeutic efficacy studies of 5 determined by assessing tumor growth and survival rate in female B10.D1 mice implanted with NT2 cells showed that out of ten mice vaccinated, seven were still alive eight weeks after tumor inoculation, whereas only one or none survived in negative control groups. Finally, in vivo depletion of B cells, CD4+ cells, or CD8+ T cells in immunized mice by using specific mAbs showed that only depletion of B cells abrogated the protection induced by 5 against tumor progression and death, suggesting that the protection is mainly due to B cells.
In the frame of the design of anticancer vaccine, the group of Danishefsky proposed a prototype displaying multiple copies of two TACAs, the Tn and STn antigens, assembled on a cyclic peptidic scaffold using a convergent synthesis.11 These two carbohydrate epitopes are over-expressed on the surfaces of a variety of epithelial cancers and are presented in broadly conserved clusters of 2–4 carbohydrate units. The cyclopeptidic scaffold contains two pairs of D-Pro-L-Pro as β-turn inducers and located opposite one another in the sequence,87 four aspartic acid residues as glycan points of attachment and one unique cysteine residue for carrier protein conjugation. The resulting cyclic 12-mer peptide conformation directs the side chain groups of the aspartic acid residues above the plane determined by the scaffold and the cysteine side chain residue projecting from the bottom (Fig. 9).
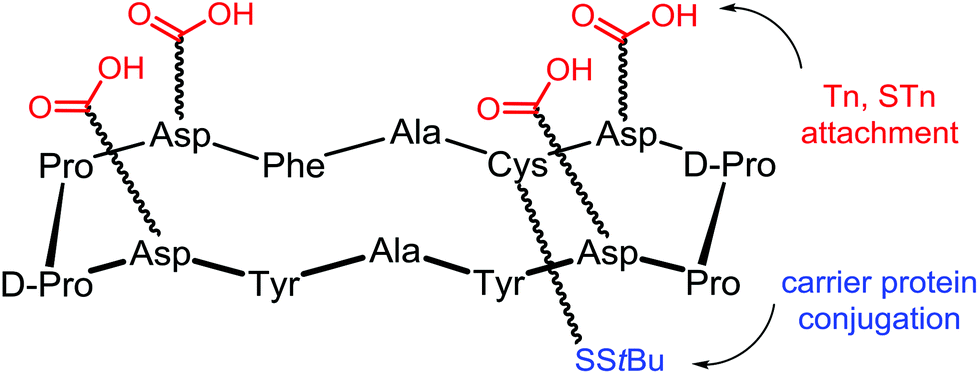 | ||
| Fig. 9 Scaffold used by Dansihefsky to attach Tn and STn antigens. | ||
The Tn and STn functionalized building blocks, respectively 7 and 8,88 were used as handles for coupling to the peptide scaffold (Fig. 10). Tetra- and hexadecavalent homo-antigenic vaccines prototypes were first constructed, compounds 9 and 10 displaying respectively four and six Tn residues and compound 11 being decorated by STn units (Fig. 10).
 | ||
| Fig. 10 Homo- and hetero-displayed Tn/STn-containing clusters synthesized by Danishefsky's group. | ||
Cyclic peptides containing six or four aspartate residues, for accessing to compounds 9–11, were prepared through automated solid-phase synthesis; after tert-butyl ester deprotection building blocks 7 or 8 were coupled under conditions known to minimize aspartimide formation.89–93 Next, Danishefsky and coworkers turned to the synthesis of the unimolecular multi-hetero-antigenic construct, 12, wherein both Tn and STn antigens are displayed on the cyclopeptidic scaffold. This type of clusters is intended to reflect the degree of carbohydrate heterogeneity associated with most cancers.94,95 It is postulated that such hetero-glyco-clusters could activate different populations of B cells to produce Abs with multiple selectivity and that these Abs could target tumors at different stage of the disease progression. This unimolecular multi-epitope construct was obtained following a sequential attachment of building blocks 7 and 8 onto a scaffold bearing orthogonal protected aspartate residues and subsequent global deacetylation. Another original feature of this work concerns the utilization of ring-closing metathesis (RCM) to incorporate an internal cross-linking element between carbohydrates, in order to rigidify the construct and minimize the spreading of glycan units. This type of constrained architecture is expected to better mimic tightly clustered B cell carbohydrate epitope and thus increase the antibody response. Construct 13 has been obtained from intermediate 14 performing a RCM reaction followed by reduction of the resulting olefin96 was found to give good yield and avoid terminal olefin isomerisation as well (Scheme 1).
 | ||
| Scheme 1 Constrained Tn-bearing cluster synthesized by Danishefsky and co-workers. | ||
However, immunological investigations of these promising constructs have not been reported yet.
Anti-viral vaccine design
A few studies describing the utilization of cyclopeptide scaffolds as carrier in the design of vaccines against viruses have been reported. For example, Danishefsky and co-workers have synthesis of carbohydrate-based anti-HIV vaccine prototypes.97 Only few antibodies have been identified to bind the heavily glycosylated exposed faces of the HIV envelope spike protein gp120. Among them, the human 2G12 antibody was found to neutralize a broad range of HIV strains by interacting with the Man9GlcNAc2 epitope expressed in the silent region of gp120 glycoprotein. Several experiments98,99 including a co-crystal structure100,101 showed that clusters of the high-mannose glycan moiety 15 can bind to 2G12 efficiently (Fig. 11). Although synthetic structures containing several copies of the glycan 15 have been already synthesized along with the proximal portion of a gp120 amino acid sequence, no affinity for 2G12 has been observed. Danishefsky and co-workers suggested that the peptide component should present the glycans in a suitable orientation to ensure recognition. | ||
| Fig. 11 Structure of high-mannose oligosaccharide epitope Man9GlcNAc2. | ||
To confirm this hypothesis, they chose a 14-residue cyclic peptide scaffold to achieve much greater design flexibility compared to the natural sequence (Fig. 12). This platform allowed for variation in the number of glycan attachments and distances between glycans units. Moreover, the presence in this scaffold of a chemical handle, spatially oriented in the opposite side of glycan units, provided a suitable anchorage point for conjugation to a carrier protein for further immunological assays. After conversion of fully deprotected glycan moiety 15 to glycosyl amine 16 by Kotchetkov amination,102 peptidic scaffolds bearing two to three aspartate residues were reacted to give the double and triple aspartylation products, respectively 17 and 18.
 | ||
| Fig. 12 2G12-epitope mimics by Danishefsky and co-workers. | ||
The binding properties of these construct were evaluated by surface plasmon resonance on a freshly prepared 2G12 surface with the non- and mono-glycosylated peptides as controls. Only divalent and trivalent glycopeptides 17 and 18 showed strong binding to the 2G12 surface, highlighting the importance of multivalent interactions and suggesting homology to the natural epitope on gp120. Having proved the ability of their synthetic glycopeptides to mimic 2G12 epitope, Danishefsky and co-workers next proceeded by coupling the selected divalent construct 17 to the outer membrane protein complex (OMPC) derived from Neisseria meningitidis, a macromolecular lipoprotein complex used as the immunogenic carrier.103–105,112 For this purpose, surface-accessible lysine residues of OMPC were maleimidated and the activated carrier was reacted with unmasked thiol group of 19 to give the conjugate 20 (Scheme 2).
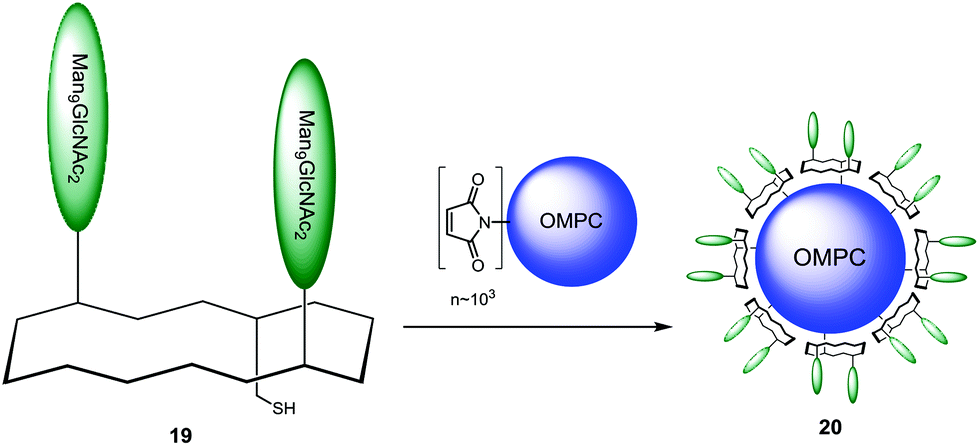 | ||
| Scheme 2 Conjugation of epitope mimic to immunogenic carrier. | ||
The thiol–maleimide coupling was controlled by quantification of dicarboxyethylcysteine produced upon acid hydrolysis. The peptide loading was found to be ranged from ∼2000 to 3000 mol peptide per mol carrier complex, indicating that ∼50% of available maleimide groups were derivatized.106 The ability of the glycoconjugate 20 to bind human 2G12 antibody was qualitatively investigated by an ELISA sandwich assay. For this, plates coated with gp160, compound 20 or OMPC alone were incubated with 2G12. HRP antihuman IgG was used as secondary antibody. The results showed that even though compound 20 gave a clear response compared to the negative control, it was still less efficient than gp160 positive control. The immunogen capacity of the glycoconjugate 20 to induce a 2G12-like neutralizing antibody response was then evaluated. Immunization of guinea pigs and rhesus macaques was shown to promote carbohydrate-specific Ab responses in both species; unfortunately these antibodies were not able to neutralize HIV strains and although compound 20 was able to bind 2G12, it was poor competitor for binding of the mAb to native gp120.
Similarly, Wang and co-workers designed a new class of template-assembled oligomannose cluster as epitope mimics for HIV-neutralizing antibody 2G12.107 A previous work, in which an oligomannose cluster Tetra-Man9 was conjugated to the keyhole limpet hemocyanin (KLH), has shown that the majority of antibody responses were directed to the linkers and the template, thus diluting drastically the immune response to the oligomannose cluster. For this reason, they decided to design new conjugate changing the carrier nature.108 Based on immunization studies that have demonstrated the non-immunogenicity of RAFT scaffold,109 they designed a novel cyclopeptide displaying the oligomannose cluster, in which four α-linked Man4 oligosaccharides corresponding to the tetramannose D1 arm of Man9GlcNAc2 epitope were introduced at one face of the scaffold, and two copies of universal T-helper epitopes corresponding to the sequence (TT830–844) of tetanus toxoid110 were introduced at the other face (Fig. 13).
 | ||
| Fig. 13 (a) Structures of Man9GlcNAc2, and (b) the designed template-assembled glycopeptide prototype vaccine. | ||
The D1 arm tetrasaccharide derivative containing an azido functionality at the aglycon portion was synthesized in two variants regarding the linker length (21 and 22), along with a fluorinated derivative of 21 (23) and the mannose arm 24 (Fig. 14).
 | ||
| Fig. 14 Azido-bearing D1 arm tetrasaccharide derivatives and mono-mannose analogue. | ||
These arms were conjugated to a tetrapropargylated scaffold using copper(I)-catalyzed azido-alkyne cycloaddition (CuAAC) to give the corresponding glycoclusters (25–28) with excellent yields (Scheme 3).
 | ||
| Scheme 3 Mannose-functionalyzed clusters as mimics of the 2G12 epitope. | ||
Finally, for the two T-helper peptides introduction, the fluorinated oligosaccharide cluster 27 was derived with two azide linkers in the lower face of the platform, to give compound 29, which underwent CuAAC reaction with the propargyl moiety previously introduced in the peptide moiety-terminus, to give the fully synthetic vaccine prototype 30 with good yields (Scheme 4).
 | ||
| Scheme 4 Conjugation of the Th epitope. | ||
The binding abilities of the obtained clusters to human 2G12 antibody were evaluated by SPR experiments.111 In agreement with the previous observation of Danishefsky and co-workers,112 it was found that at 10 μM, all the synthetic monomers 21–23, as well as the high-mannose type N-glycan Man9GlcNAc2Asn, did not show apparent binding to antibody 2G12. The template assembled mannosyl-based glycocluster 25 neither showed detectable binding, while the synthetic oligosaccharide clusters carrying four units of the D1 arm tetrasaccharide (26–28) have demonstrated apparent affinity to the antibody 2G12 in terms of response units (RU). Also the fluorinated compound 27, despite a reduced efficiency in comparison with compounds 26 and 28, exhibits significant affinity for 2G12 (43 RU). This data suggests that an appropriate distance between the scaffold and the sugar moiety allows an improvement for 2G12 antibody binding.
Finally, the 2G12 binding of the fully synthetic vaccine prototype 30, containing two T-helper peptides was examined. This compound demonstrated almost identical binding profiles and kinetic outcome to those of the corresponding fluorinated glycocluster 27, which suggests that the introduction of the T-helper moiety onto the cyclic decapeptide template did not affect the structural integrity of the oligosaccharide cluster formed at the other face of the template.
Conclusion and prospect
This article presents an overview of the utilization of cyclopeptide as scaffold for multivalent and heterogeneous presentation of carbohydrate B-cell and peptidic T-cell epitopes for the design of synthetic vaccines. We highlighted the numerous advantages offered by cyclopeptide templates for vaccine constructs: (i) its non-immunogenicity; (ii) its two independent functional faces that can be regioselectively functionalized with several carbohydrate units as B cell antigens, and the other for the conjugation of T cell peptide; (iii) the possibility to easily combine diverse epitopes; (iv) the control in the glycan composition to ensure a reproducible immune response. To date, the most sophisticated structures display four different immunogenic components that were shown to elicit efficient responses in mice models against cancer. In addition, carbohydrates based-vaccine candidates were also reported for antiviral applications and have demonstrated promising biological activities. While fully synthetic carbohydrate-based vaccine candidates have not been yet evaluated in clinical trial, recent promising results undoubtedly open new perspectives for immunotherapy to promote either for therapeutic or prophylactic effects. Due to their synthetic modularity, cyclopeptides seem ideal to combine several carbohydrate antigens on the same vaccine prototype in order to promote a multifaceted immune response against different types of cancers, and to respond to a wider population of tumor cells and at different stage of the disease progression. Other structural modifications such as the presentation of different display and/or higher density of carbohydrate epitopes, of combinations of different antigenic peptides and of glycopeptide mucin fragments represent interesting directions for future development towards immunological optimization.Acknowledgements
Part of these studies was supported by CNRS, Université Grenoble Alpes, the “Communauté d'agglomération Grenoble-Alpes Métropole” (Nanobio program) and Labex ARCANE (ANR-11-LABX-0003-01). O. R. acknowledge, the French Agence Nationale de la Recherche (ANR-12-JS07-0001-01 “VacSyn”) and the European Research Council Consolidator Grant “LEGO” (647938).References
- B. Ernst, W. Hart and P. Sinay, Carbohydrates in Chemistry and Biology, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- F. Peri, Chem. Soc. Rev., 2013, 42, 4543–4556 RSC.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- P. Sears and C.-H. Wong, Science, 2001, 291, 2344–2350 CrossRef CAS PubMed.
- N. Gaidzik, U. Westerlind and H. Kunz, Chem. Soc. Rev., 2013, 42, 4421–4442 RSC.
- D. P. Galonic and D. Y. Gin, Nature, 2007, 446, 1000–1007 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed.
- C. M. Snapper and J. J. Mond, J. Immunol., 1996, 157, 2229–2233 CAS.
- W. M. Kalka-Moll, A. O. Tzianabos, P. W. Bryant, M. Niemeyer, H. L. Ploegh and D. L. Kasper, J. Immunol., 2002, 169, 6149–6153 CrossRef CAS.
- O. T. Avery and W. F. Goebel, J. Exp. Med., 1929, 50, 521–533 CrossRef.
- I. Jeon, D. Lee, I. J. Krauss and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 14337–14344 CrossRef CAS PubMed.
- M. A. Johnson and D. R. Bundle, Chem. Soc. Rev., 2013, 42, 4327–4344 RSC.
- J. D. Warren, X. Geng and S. J. Danishefsky, Top. Curr. Chem., 2007, 267, 109–141 CrossRef CAS.
- L. Morelli, L. Poletti and L. Lay, Eur. J. Org. Chem., 2011, 5723–5777 CrossRef CAS.
- K. L. O′Brien, M. C. Steinhoff, K. Edwards, H. Keyserling, M. L. Thoms and D. Madore, Pediatr. Infect. Dis. J., 1996, 15, 425–430 CrossRef.
- H. Kayhty, J. Eskola, H. Peltola, L. Saarinen and P. H. Makela, J. Infect. Dis., 1992, 165, S165–S166 CrossRef PubMed.
- A. Leach, P. A. Twumasi, S. Kumah, W. S. Banya, S. Jaffar, B. D. Forrest, D. M. Granoff, D. E. LiButti, G. M. Carlone, L. B. Pais, C. V. Broome and B. M. Greenwood, J. Infect. Dis., 1997, 175, 200–204 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed.
- D. Feng, A. S. Shaikh and F. Wang, ACS Chem. Biol., 2016, 11, 850–863 CrossRef CAS PubMed.
- T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem., Int. Ed., 2011, 50, 2–24 CrossRef PubMed.
- J. P. Richardson and D. Macmillan, in Glycoscience, ed. B. Fraser-Reid, K. Tatsuta and J. Thiem, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed.
- H. Cai, Z.-H. Huang, L. Shi, Z.-Y. Sun, Y.-F. Zhao, H. Kunz and Y.-M. Li, Angew. Chem., Int. Ed., 2012, 51, 1719–1723 CrossRef CAS PubMed.
- E. M. Pollabauer, R. Petermann and H. J. Ehrlich, Vaccine, 2009, 27, 1674–1679 CrossRef PubMed.
- R. Dagan, J. Eskola, C. Leclerc and O. Leroy, Infect. Immun., 1998, 66, 2093–2098 CAS.
- R. A. Insel, Ann. N. Y. Acad. Sci., 1995, 754, 35–67 CrossRef CAS PubMed.
- L. A. Herzenberg and T. Tokuhisa, Nature, 1980, 285, 664–667 CrossRef CAS PubMed.
- M. P. Schutze, E. Deriaud, G. Przewlocki and C. Leclerc, J. Immunol., 1989, 142, 2635–2640 CAS.
- X. Renjifo, S. Wolf, P. P. Pastoret, H. Bazin, J. Urbain and O. Leo, J. Immunol., 1998, 161, 702–706 CAS.
- L. A. Herzenberg and T. Tokuhisa, Exp. Med., 1982, 155, 1730–1740 CrossRef CAS.
- J. C. Reece, H. M. Geysen and S. J. Rodda, J. Immunol., 1993, 151, 6175–6184 CAS.
- S. Bay, R. Lo-Man, E. Osinaga, H. Nakada, C. Leclerc and D. Cantacuzène, J. Pept. Res., 1997, 49, 620–625 CrossRef CAS PubMed.
- S. Grigalevicius, S. Chierici, O. Renaudet, R. Lo-Man, E. Dériaud, C. Leclerc and P. Dumy, Bioconjugate Chem., 2005, 16, 1149–1159 CrossRef CAS PubMed.
- P. Dumy, I. M. Eggleston, S. Cervigni, U. Silla, X. Sun and M. Mutter, Tetrahedron Lett., 1995, 36, 1255–1258 CrossRef CAS.
- P. Dumy, I. M. Eggleston, G. Esposito, S. Nicula and M. Mutter, Biopolymers, 1996, 39, 297–308 CrossRef CAS PubMed.
- S. Peluso, T. Rückle, C. Lehmann, M. Mutter, C. Peggion and M. Crisma, ChemBioChem, 2001, 2, 432–437 CrossRef CAS PubMed.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis, A Practical Approach, Oxford University Press, 2000 Search PubMed.
- B. Thomas, N. Berthet, J. Garcia, P. Dumy and O. Renaudet, Chem. Commun., 2013, 49, 10796–10798 RSC.
- G. C. Daskhan, N. Berthet, B. Thomas, M. Fiore and O. Renaudet, Carbohydr. Res., 2015, 405, 12–22 CrossRef PubMed.
- N. Berthet, B. Thomas, I. Bossu, E. Gillon, N. Spinelli, A. Imberty, P. Dumy and O. Renaudet, Bioconjugate Chem., 2013, 24, 1598–1611 CrossRef CAS PubMed.
- C. Pifferi, D. Goyard, E. Gillon, A. Imberty and O. Renaudet, ChemPlusChem, 2017 DOI:10.1002/cplu.201600569.
- M. C. Galan, P. Dumy and O. Renaudet, Chem. Soc. Rev., 2013, 42, 4599–4612 RSC.
- T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem., Int. Ed., 2011, 50, 1770–1791 CrossRef CAS PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed.
- R. Lo-Man, S. Bay, S. Vichier-Guerre, E. Dériaud, D. Cantacuzéne and C. Leclerc, Cancer Res., 1999, 59, 1520–1524 CAS.
- D. Boturyn, J.-L. Coll, E. Garanger, M.-C. Favrot and P. Dumy, J. Am. Chem. Soc., 2004, 126, 5730–5739 CrossRef CAS PubMed.
- O. Renaudet and P. Dumy, Org. Lett., 2003, 5, 243–246 CrossRef CAS PubMed.
- Y. Singh, O. Renaudet, E. Defrancq and P. Dumy, Org. Lett., 2005, 7, 1359–1362 CrossRef CAS PubMed.
- K. Rose, J. Am. Chem. Soc., 1994, 116, 30–33 CrossRef CAS.
- J. Shao and P. Tam, J. Am. Chem. Soc., 1995, 117, 3893–3899 CrossRef CAS.
- L. E. Canne, P. Botti, R. J. Simon, Y. Chen, E. A. Dennis and S. B. H. Kent, J. Am. Chem. Soc., 1999, 121, 8720–8727 CrossRef CAS.
- J. Katajisto, P. Virta and H. Loennberg, Bioconjugate Chem., 2004, 15, 890–896 CrossRef CAS PubMed.
- P. Neuner, P. Gallo, L. Orsatti, L. Fontana and P. Monaci, Bioconjugate Chem., 2003, 14, 276–281 CrossRef CAS PubMed.
- E. C. Rodriguez, K. A. Winans, D. S. King and C. R. Bertozzi, J. Am. Chem. Soc., 1997, 119, 9905–9906 CrossRef CAS.
- S. E. Cervigni, P. Dumy and M. Mutter, Angew. Chem., Int. Ed. Engl., 1996, 35, 1230–1232 CrossRef CAS.
- D. Forget, D. Boturyn, E. Defrancq, J. Lhomme and P. Dumy, Chem. – Eur. J., 2001, 7, 3976–3984 CrossRef CAS.
- J. Chen, W. Zeng, R. Offord and K. Rose, Bioconjugate Chem., 2003, 14, 614–618 CrossRef CAS PubMed.
- G.-A. Cremer, N. Bureaud, D. Lelievre, V. Piller, F. Piller and A. Delmas, Chem. – Eur. J., 2004, 10, 6353–6360 CrossRef CAS PubMed.
- W. Zeng, D. C. Jackson, J. Murray, K. Rose and L. E. Brown, Vaccine, 2000, 18, 1031–1039 CrossRef CAS PubMed.
- K. Rose, W. Zeng, L. E. Brown and D. C. Jackson, Mol. Immunol., 1995, 32, 1031–1037 CrossRef CAS PubMed.
- W. Zeng, S. Ghosh, M. Macris, J. Pagnon and D. C. Jackson, Vaccine, 2001, 19, 3843–3852 CrossRef CAS PubMed.
- L. Cipolla, M. Rescigno, A. Leone, B. La Ferla and F. Nicotra, Bioorg. Med. Chem., 2002, 10, 1639–1646 CrossRef CAS PubMed.
- S. Vichier-Guerre, R. Lo-Man, V. Huteau, E. Dériaud, C. Leclerc and S. Bay, Bioorg. Med. Chem. Lett., 2004, 14, 3567–3570 CrossRef CAS PubMed.
- S. J. Keding, A. Endo and S. J. Danishefsky, Tetrahedron, 2003, 59, 7023–7031 CrossRef CAS.
- G. Ragupathi, D. M. Coltard, L. J. Williams, F. Koide, E. Kagan, J. Allen, C. Harris, P. W. Glunz, P. O. Livongston and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13699–13704 CrossRef CAS PubMed.
- R. Lo-Man, S. Bay, S. Vichier-Guerre, E. Dériaud, D. Cantacuzène and C. Leclerc, Cancer Res., 1999, 59, 1520–1524 CAS.
- L. Cipolla, M. Rescigno, A. Leone, B. La Ferla and F. Nicotra, Bioorg. Med. Chem., 2002, 10, 1639–1646 CrossRef CAS PubMed.
- L. BenMohamed, G. Bertrand, C. D. McNamara, H. Gras-Masse, J. Hammer, S. L. Wechsler and A. B. Nesburn, J. Virol., 2003, 77, 9463–9473 CrossRef CAS PubMed.
- A. J. Zajac, K. Murali-Krishna, J. N. Blattman and R. Ahmed, Curr. Opin. Immunol., 1998, 10, 444–449 CrossRef CAS PubMed.
- X. Zhu, T. V. Ramos, H. Gras-Masse, B. E. Kaplan and L. BenMohamed, Eur. J. Immunol., 2004, 34, 1142–1149 CrossRef PubMed.
- J. E. Blaney, E. Nobusawa, M. A. Brehm, R. H. Bonneau, L. M. Mylin, T. M. Fu, Y. Kawaoka and S. Tevethia, J. Virol., 1998, 72, 9567–9574 CAS.
- K. M. Khanna, A. J. Lepisto and R. L. Hendricks, Trends Immunol., 2004, 25, 230–234 CrossRef CAS PubMed.
- Z. Mikloska, M. Rucckholdt, I. Ghadiminejad, H. Dunckley, M. Denis and A. L. Cunningham, J. Immunol., 2000, 164, 5167–5176 CrossRef CAS.
- K. S. Rosenthal, H. Mao, W. I. Horne, C. Wright and D. Zimmerman, Vaccine, 1999, 17, 535–542 CrossRef CAS PubMed.
- A. A. Chentoufi, A. B. Nesburn and L. BenMohamed, Arch. Immunol. Ther. Exp., 2009, 57, 409–423 CrossRef CAS PubMed.
- L. BenMohamed, R. Krishnan, C. Auge, J. F. Primus and D. J. Diamond, Immunology, 2002, 106, 113–121 CrossRef CAS PubMed.
- L. BenMohamed, R. Krishnan, J. Longmate, C. Auge and L. Low, Hum. Immunol., 2000, 61, 764–777 CrossRef CAS PubMed.
- L. BenMohamed, A. Thomas, M. Bossus, K. Brahimi and J. Wubben, Vaccine, 2000, 18, 2843–2855 CrossRef CAS PubMed.
- L. BenMohamed, A. Thomas and P. Druilhe, Infect. Immun., 2004, 72, 4376–4384 CrossRef CAS PubMed.
- C. Bourgeois and C. Tanchot, Eur. J. Immunol., 2003, 33, 3225–3231 CrossRef CAS PubMed.
- S. J. Keding and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11937–11942 CrossRef CAS PubMed.
- V. Kudryashov, P. W. Glunz, L. J. Williams, S. Hintermann and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3264–3269 CrossRef CAS PubMed.
- O. Renaudet, L. BenMohamed, G. Dasgupta, I. Bettahi and P. Dumy, ChemMedChem, 2008, 2, 425–431 Search PubMed.
- O. Rötzschke, K. Falk, S. Stevanovic, G. Jung, P. Walden and H.-G. Rammen, Eur. J. Immunol., 1991, 21, 2891–2894 CrossRef PubMed.
- J. Alexander, M.-F. del Guercio, A. Maewal, L. Qia, J. Fikes, R. W. Chesnut, J. Paulson, D. R. Bundle, S. Defrees and A. Sette, J. Immunol., 2000, 164, 1625–1633 CrossRef CAS.
- B. Richichi, B. Thomas, M. Fiore, R. Bosco, H. Qureshi, C. Nativi, O. Renaudet and L. BenMohamed, Angew. Chem., Int. Ed., 2014, 53, 11917–11920 CrossRef CAS PubMed.
- J. Jiménez-Barbero, E. Dragoni, C. Venturi, F. Nannucci, A. Ardà, M. Fontanella, S. André, F. Javier Caçada, H.-J. Gabius and C. Nativi, Chem. – Eur. J., 2009, 15, 10423–10431 CrossRef PubMed.
- M. Favre, K. Moehle, J. Jiang, B. Pfeiffer and J. Robinson, J. Am. Chem. Soc., 1999, 121, 2679–2685 CrossRef CAS.
- S. J. Keding, A. Endo and S. J. Danishefsky, Tetrahedron Lett., 2003, 4, 3413–3416 CrossRef.
- M. Bodanszky and S. Natarajan, J. Org. Chem., 1975, 40, 2495–2249 CrossRef CAS PubMed.
- M. Bodanszky and J. Z. Kwei, Int. J. Pept. Protein Res., 1978, 12, 69–74 CrossRef CAS PubMed.
- J. P. Tam, M. W. Riemen and R. B. Merrifield, Pept. Res., 1988, 1, 6–18 CAS.
- M. Mergler, F. Dick, B. Sax, P. Weiler and T. Vorherr, J. Pept. Sci., 2003, 9, 36–46 CrossRef CAS PubMed.
- J. L. Lauer, C. G. Fields and G. B. Fields, Lett. Pept. Sci., 1994, 1, 197–205 CrossRef.
- S. L. Zhang, C. Cordon-Cardo, H. S. Zhang, V. E. Reuter, S. Adluri, W. B. Hamilton, K. O. Lloyd and P. O. Livingston, Int. J. Cancer, 1997, 73, 42–49 CrossRef CAS PubMed.
- S. L. Zhang, H. S. Zhang, C. Cordon-Cardo, V. E. Reuter, A. K. Singhal, K. O. Lloyd and P. O. Livingston, Int. J. Cancer, 1997, 73, 50–56 CrossRef CAS PubMed.
- J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791–799 CrossRef CAS.
- I. J. Krauss, J. G. Joyce, A. C. Finnefrock, H. C. Song, V. Y. Dudkin, X. Geng, J. D. Warren, M. Chastain, J. W. Shiver and S. J. Danishefsky, J. Am. Chem. Soc., 2007, 129, 11042–11044 CrossRef CAS PubMed.
- C. N. Scanlan, R. Pantophlet, M. R. Wormald, E. O. Saphire, R. Stanfield, I. A. Wilson, H. Katinger, R. A. Dwek, P. M. Rudd and D. R. Burton, J. Virol., 2002, 76, 7306–7321 CrossRef CAS PubMed.
- R. W. Sanders, M. Venturi, L. Schiffner, R. Kalyanaraman, H. Katinger, K. O. Lloyd, P. D. Kwong and J. P. Moore, J. Virol., 2002, 76, 7293–7305 CrossRef CAS PubMed.
- D. A. Calarese, C. N. Scanlan, M. B. Zwick, S. Deechongkit, Y. Mimura, R. Kunert, P. Zhu, M. R. Wormald, R. L. Stanfield, K. H. Roux, J. W. Kelly, P. M. Rudd, R. A. Dwek, H. Katinger, D. R. Burton and I. A. Wilson, Science, 2003, 300, 2065–2071 CrossRef CAS PubMed.
- D. A. Calarese, H.-K. Lee, C.-Y. Huang, M. D. Best, R. D. Astronomo, R. L. Stanfield, H. Katinger, D. R. Burton, C.-H. Wong and I. A. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13372–13377 CrossRef CAS PubMed.
- L. M. Likhosherstov, O. S. Novikova, V. A. Derevitskaja and N. K. Kochetkov, Carbohydr. Res., 1986, 146, C1–C5 CrossRef CAS.
- G. B. McGaughey, M. Citron, R. C. Danzeisen, R. M. Freidinger, V. M. Garsky, W. M. Hurni, J. G. Joyce, X. Liang, M. Miller, J. Shiver and M. J. Bogusky, Biochemistry, 2003, 42, 3214–3223 CrossRef CAS PubMed.
- J. G. Joyce, C. Abeygunawardana, Q. Xu, J. C. Cook, R. Hepler, C. T. Przysiecki, K. M. Grimm, K. Roper, C. C. Yu Ip, L. Cope, D. Montgomery, M. Chang, S. Campie, M. Brown, T. B. McNeely, J. Zorman, T. Maira-Litran, G. B. Pier, P. M. Keller, K. U. Jansen and G. E. Mark, Carbohydr. Res., 2003, 338, 903–922 CrossRef CAS PubMed.
- J. Joyce, J. Cook, D. Chabot, R. Hepler, W. Shoop, Q. Xu, T. Stambaugh, M. Aste-Amezaga, S. Wang, L. Indrawati, M. Bruner, A. Friedlander, P. Keller and M. Caulfield, J. Biol. Chem., 2006, 281, 4831–4843 CrossRef CAS PubMed.
- J. G. Joyce, I. J. Krauss, H. C. Song, D. W. Opalka, K. M. Grimm, D. D. Nahas, M. T. Esser, R. Hrin, M. Feng, V. Y. Dudkin, M. Chastain, J. W. Shiver and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15684–15689 CrossRef CAS PubMed.
- J. Wang, H. Li, G. Zou and L.-X. Wang, Org. Biomol. Chem., 2007, 5, 1529–1540 CAS.
- J. Ni, H. Song, Y. Wang, N. M. Stamatos and L.-X. Wang, Bioconjugate Chem., 2006, 17, 493–500 CrossRef CAS PubMed.
- S. Grigalevicius, S. Chierici, O. Renaudet, R. Lo-Man, E. Deriaud, C. Leclerc and P. Dumy, Bioconjugate Chem., 2005, 16, 1149–1159 CrossRef CAS PubMed.
- P. Panina-Bordignon, A. Tan, A. Termijtelen, S. Demotz, G. Corradin and A. Lanzavecchia, Eur. J. Immunol., 1989, 19, 2237–2242 CrossRef CAS PubMed.
- V. Y. Dudkin, M. Orlova, X. Geng, M. Mandal, W. C. Olson and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 9560–9562 CrossRef CAS PubMed.
- I. J. Krauss, J. G. Joyce, A. C. Finnefrock, H. C. Song, V. Y. Dudkin, X. Geng, J. D. Warren, M. Chastain, J. W. Shiver and S. J. Danishefsky, J. Am. Chem. Soc., 2007, 129, 11042–11044 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |