
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer brushes based on PLLA-b-PEO colloids for the preparation of protein resistant PLA surfaces†
E. Mázl Chánová‡
 *,
O. Pop-Georgievski‡,
M. M. Kumorek,
O. Janoušková,
L. Machová,
D. Kubies and
F. Rypáček
*,
O. Pop-Georgievski‡,
M. M. Kumorek,
O. Janoušková,
L. Machová,
D. Kubies and
F. Rypáček
Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic. E-mail: chanova@imc.cas.cz; Tel: +420-296809453
First published on 25th April 2017
Abstract
In this study we investigate the formation of protein-resistant polymer surfaces, such as aliphatic polyesters, through the deposition of self-assemblies of amphiphilic poly(L-lactide)-b-poly(ethylene oxide), PLLA-b-PEO, copolymers as stable nanoparticles with a kinetically frozen PLLA core on model PLLA surfaces. The length of the PEO chains in the corona was tuned to achieve polymer brushes capable of preventing protein adsorption on PLA-based biomaterials. The spectroscopic ellipsometry, IR and XPS analysis, contact angle goniometry, and AFM proved that the PEO chains adopted a brush structure and were preferably exposed on the surface. The low-fouling properties of the physisorbed PLLA-b-PEO layers approached the ones of reactive grafting methods, as shown by surface plasmon resonance spectroscopy. The anti-fouling properties of the prepared PEO brushes provided sufficient interface to prevent cell adhesion as proved in vitro. Thus, the developed surface coating with PLLA-b-PEO colloids can provide an anti-fouling background for the creation of nanopatterned biofunctionalized surfaces in biomedical applications.
Introduction
Non-fouling surfaces have been under frequent investigation for biomedical applications such as biosensing or tissue engineering.1 Current trends in the area of polymer biomaterials for tissue engineering are focused on controlled cell–surface interactions through surface functionalization of biomaterials using the bioactive motifs derived from natural structures of the extracellular matrix (such as fibrinogen or laminin) present on the surface of the biomaterial.2,3 However, the limiting factor of this approach is non-specific protein adsorption, which occurs at the first instant when the artificial material is brought into contact with body fluids or cell culture medium.4 In such a case, the accessibility of bioactive groups for expected surface–cell interactions would be restricted by the adsorbed proteins.Modification with hydrophilic polymers was reported as an efficient method for surface passivation due to the effect of steric repulsion.4 For that purpose, besides other polymers, such as polybetaine-based polymers1 or poly(vinyl alcohol),5 poly(ethylene oxide), PEO, has been extensively studied and proved to be effective.6,7 In general, the efficiency of the surface coating in preventing protein adsorption is attributed to the arrangement of the hydrophilic polymer chains into a brush conformation. The description of the theoretical model of polymer brushes and their mechanism and efficiency in suppressing the adsorption of proteins was reported by Szleifer8,9 and Halperin,10,11 and the important parameters of a polymer brush, such as chain distance, chain density, and the thickness of a polymer brush, have been established.
Several strategies based on both physical entrapment and covalent attachment of PEO and their efficiency have been reported.6,12 The appropriate method for PEO immobilization depends on the character of the original surface to be modified and the required application. Ideally, for tissue engineering applications, besides the homogeneity and stability of a non-fouling layer over the whole biomaterial surface, the layer enables the controlled incorporation of bioactive molecules. This can be accomplished by the application of a certain portion of end-functionalized PEO chains through which the surface can be modified.
Modification of surfaces via covalent immobilization provides stable PEO layers. The “grafting to” and “grafting from” techniques provided surfaces with a different density of the tethered EO based chains.6 Recently, the formation of end-tethered PEO brushes on polydopamine-modified surfaces, thus applicable virtually to any substrate, prepared by the “grafting to” method and with a significantly reduced protein adsorption has been reported.7 However, aliphatic polyesters, such as polylactide (PLA), which is a biodegradable polymer widely used for biomedical applications exhibiting a rather hydrophobic character which induces the non-specific protein adsorption, suffer from the lack of suitable functional groups for a direct modification with PEO as well as with bioactive compounds. Thus, for guided cell–surface interactions in the area of tissue engineering, physically-based surface modifications of PLLA by entrapment of PEO chains13 or deposition of PEO-based copolymers have been mostly investigated.14–16
In our approach, the deposition of well-defined colloidal nanoparticles of amphiphilic block copolymers composed of amorphous poly(DL-lactide) and semi-crystalline poly(ethylene oxide), PDLLA-b-PEO, has been studied as a method of surface modification of PLLA-based biomaterials.14 The PLLA substrates were coated with a mixture of non-functionalized and functionalized copolymers with biotin representing a model functional group. Taking advantage of specific avidin–biotin interactions, the accessibility of biotinylated PEO chains in the topmost copolymer layer was demonstrated by AFM. In a follow-up study, we demonstrated that the surface topography of individual functional groups at the end of the PEO block in such copolymer layers can be detected through the monitoring of local mechanical properties by AFM of a larger marker such as streptavidin-modified gold nanoparticles (diam. 40 nm) bound to biotin moieties on the surface.17
Smart biomaterials with well-defined distances between the peptide ligands on the surface seem to be the favorable approach in designing biomimetic surfaces for controlled cell–surface interactions. The effective ligand spacing of adhesion peptides on a nanometer scale required for cell adhesion and cell guidance has been established on model surfaces by Spatz and Cavalcanti-Adam.18–20 We believe that the one of the ways to transfer this proposed concept to real 3D biomaterials and scaffolds is colloidal printing of functional copolymers, such as PLA-b-PEO, for preparation of specific surface patterns of biomimetic groups that would influence cell adhesion, growth, and differentiation. The feasibility of our approach to prepare even-patterned surfaces was proved in our recent study.21 Different mixtures of non-functionalized and maleimide-functionalized 7/10 PLLA-b-PEO copolymers were spin-cast on the PLLA substrate from the selective solvent, fixed, and grafted with a RGD-containing oligopeptide flanked with biotin. We demonstrated that in such a way the surfaces with (i) randomly distributed RGD-peptides or with (ii) a specific distribution of RGD-peptides in clusters can be prepared. Moreover, pilot cell culture experiments with the MG63 osteosarcoma cell line proved that the surfaces with RGD-peptides arranged in clusters supported cell adhesion and spreading.21
Based on our previous studies, we have hypothesized that when using PLA-b-PEO block copolymers with appropriate molecular parameters together with a suitable selective solvent, the formed copolymer self-assemblies can provide nanoparticles with a polylactide core which is stabilized but still not completely in a glassy state. We assume that the deposition of such physically stabilized nanoparticles occurs according to Fig. 1A and results in a nanostructured surface. Taking advantage of the affinity of the PLLA chains in the nanoparticle core to the PLLA chains on the surface, we propose that the nanoparticle can relax on the surface according to the Ligoure model;22 and, hence, the PLLA chains from the core will be entangled with the PLLA chains on the original surface, thus stabilizing the coating layer. Furthermore, the PEO phase can be exposed to the outer environment, and thus the PEO chains of the nanoparticle will be present in the top layer. When a portion of the PEO chains carries a functional group for grafting of RGD-peptides, the distance between the peptides can be tuned by the size of the deposited PLLA-b-PEO nanoparticles, which is dependent on molecular parameters such as molecular weight.
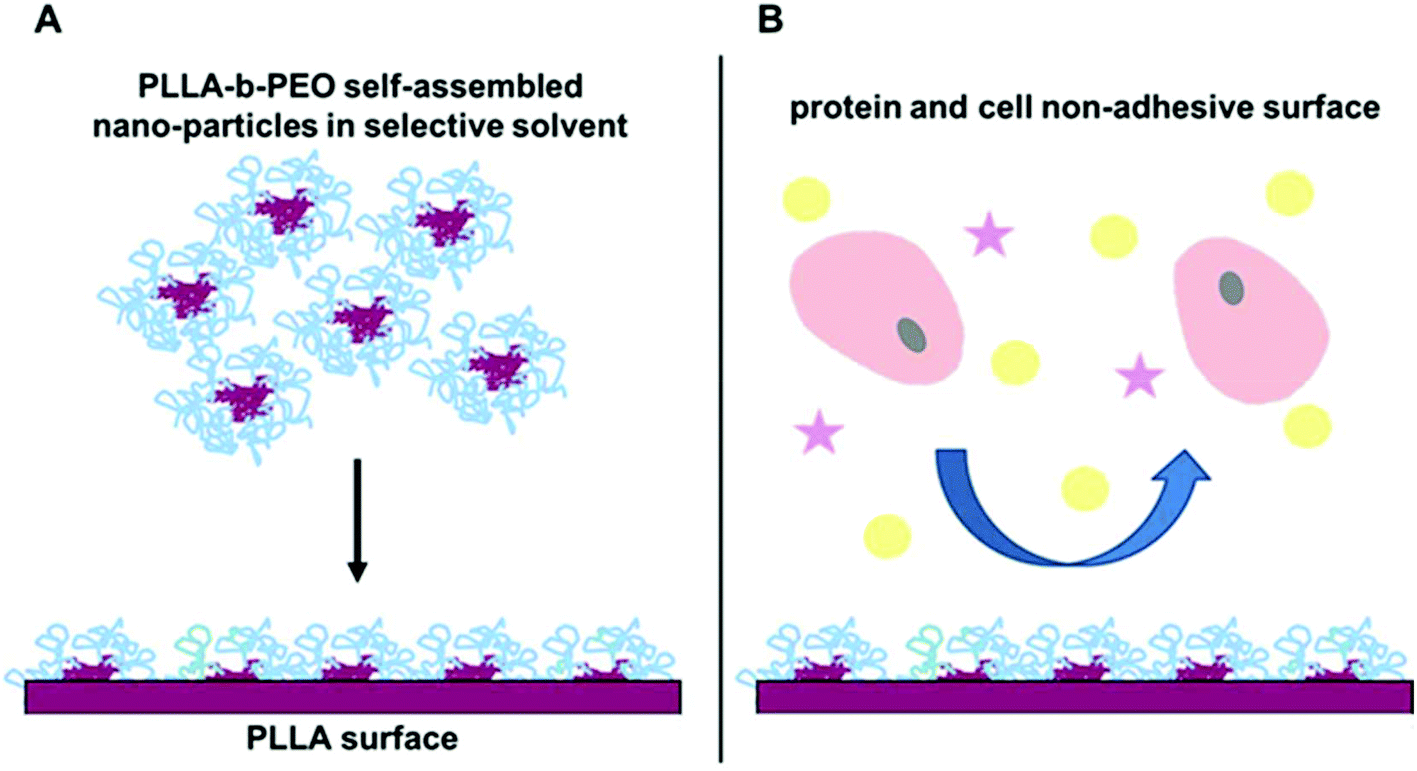 | ||
| Fig. 1 Schematic illustration of deposition of PLLA-b-PEO colloidal nanoparticles from a selective solvent onto the polylactide surface (A) for preparation of protein and cell non-adhesive surfaces (B). | ||
However, in order to study and, eventually, to control guided cell–surface interactions through the integrin-RGD mediated pathway it is necessary to eliminate any non-specific interactions mediated by spontaneous adsorption of proteins from the media. Therefore, in the present study we investigated in more detail the anti-fouling properties of surfaces modified by deposition of the PLLA-b-PEO copolymers and their dependence on the molecular parameters of the copolymers. We used three PLLA-b-PEO copolymers with a fixed length of the PLLA blocks and a variable length of the PEO blocks from 5 kDa to 15 kDa. We hypothesize that these PLLA-b-PEO copolymers with different lengths of PEO blocks will form nanoparticles with different sizes. Their deposition will result in copolymer surfaces with a different wettability, surface structure, and polymer brush formation. Furthermore, we have investigated the efficiency of the PLLA-b-PEO coatings in preventing the adsorption of single plasma proteins and proteins from cell cultivation media, and the resistance of the coatings to cell adhesion according to Fig. 1B. Finally, we correlated the results with the molecular parameters of the used copolymers.
Experimental
Materials
Acetone, toluene, methanol, diethyl ether, tetrahydrofuran (THF) (LachNer), dopamine hydrochloride (Aldrich), human serum albumin (HSA, Aldrich), fibrinogen (Fbg, Aldrich), Minimum Essential Medium Eagle (MEME, Aldrich), Human Umbilical Vein Endothelial Cells (HUVEC, Life technologies), Medium 200 and low serum growth supplements (Life Technologies), human osteosarcoma cell line MG63 (Aldrich), fetal bovine serum (FBS, Aldrich), glutamine (Aldrich), non-essential amino acids (Aldrich), penicillin and streptomycin (Life Technologies), trypsin (Aldrich) and α-hydroxy-ω-methoxy-poly(ethylene oxide) Mw = 5, 10, and 15 kDa (Shearwater Nektar) were used as purchased.Tin(II) 2-ethylhexanoate (Sn(Oct)2) (Aldrich) was distilled under vacuum and stored under an inert gas at −18 °C. The monomer L-lactide (Aldrich) was used freshly recrystallized from a mixture of ethyl acetate and toluene.
Methods
First, the substrates (both SPR Au chips and glass slides) were cleaned and modified with a PDA layer by a standard protocol as described in our previous studies.7,23
Then, the PLLA film was prepared by spin-casting from the polylactide solution in chloroform (4 mg ml−1, 180 μl on each SPR substrate, 60 μl on each glass slide) using a PWM32-PS-R790 spinner (Headway Research, Inc.) at 2750 rpm for 30 s at 24 °C. The spin-cast films were annealed under vacuum at 120 °C for 3 h.
Finally, the copolymer surfaces were prepared by spin-casting from suspensions in the acetone/methanol mixed solvent (1/9 v/v, copolymer concentration: 3 mg ml−1) on the PLLA surface. Particularly, 300 μl of a given suspension was spin-cast on at least 4 SPR substrates at 25 °C for 50 s in total (step 1: 1000 rpm for 10 s, step 2: 1750 rpm for 40 s), and 70 μl on each glass slide. The copolymer layers were heated overnight at 50 °C under vacuum. Subsequently, the layers were annealed in Milli-Q water at 40 °C for 1 h and quenched by plunging into ice-cold Milli-Q water afterwards. The samples were dried at RT and stored under vacuum.
The PEO-b-PLLA thicknesses obtained from the ellipsometry measurements were used to calculate the chain density 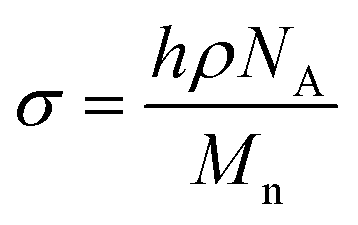 and distance between the block copolymer chains supposing a hexagonal packing
and distance between the block copolymer chains supposing a hexagonal packing 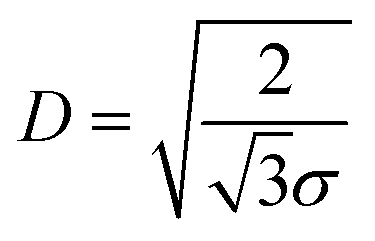 , where h is the layer thickness in the dry state as determined by ellipsometry,26–28ρ = 1.09 g cm−3 is the PEO bulk density, Mn is the number average molecular weight of the block copolymers and NA is the Avogadro constant. The ratio of the distance between the PEO block to the radius of gyration of PEO (Rg PEO), i.e.
, where h is the layer thickness in the dry state as determined by ellipsometry,26–28ρ = 1.09 g cm−3 is the PEO bulk density, Mn is the number average molecular weight of the block copolymers and NA is the Avogadro constant. The ratio of the distance between the PEO block to the radius of gyration of PEO (Rg PEO), i.e.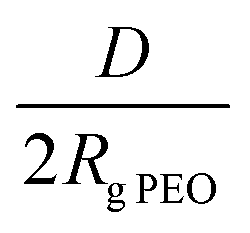 describes the overlapping of the tethered chains. Values of
describes the overlapping of the tethered chains. Values of 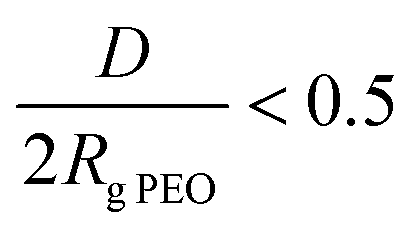 indicate that the polymer chains are in a “brush” state. The Rg of PEO in water was calculated according to Kawaguchi et al.29
indicate that the polymer chains are in a “brush” state. The Rg of PEO in water was calculated according to Kawaguchi et al.29
The analyzer transmission function, Scofield sensitivity factors, and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2 M formalism. The BE scale of the XPS spectrometer was calibrated by the well-known positions of the C–C and C–H, C–O and C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O peaks of polyethylene terephthalate and Cu 2p, Ag 3d, and Au 4f peaks of metallic Cu, Ag and Au, respectively. All measured spectra were charge referenced to the C 1s peak attributed to C–C, C–H at 285.0 eV binding energy. The obtained high resolution spectra were fitted with Voigt profiles to probe the individual contributions of present chemical species. The resulting binding energy uncertainty of the reported measurements and analysis did not exceed ±0.3 eV.
O)–O peaks of polyethylene terephthalate and Cu 2p, Ag 3d, and Au 4f peaks of metallic Cu, Ag and Au, respectively. All measured spectra were charge referenced to the C 1s peak attributed to C–C, C–H at 285.0 eV binding energy. The obtained high resolution spectra were fitted with Voigt profiles to probe the individual contributions of present chemical species. The resulting binding energy uncertainty of the reported measurements and analysis did not exceed ±0.3 eV.
The samples with the cells were incubated under a 5% CO2 atmosphere at 37 °C. The cell adhesion and growth were evaluated after 1, 3, and 5 days using an OPTIKA inverted microscope and by determination of cell number using the Alamar Blue viability assay according to Kasoju et al.31 Briefly, the slides with prepared surfaces were transferred to a new 24-well plate with 300 μl of medium and 30 μl of Alamar Blue (AB) cell viability reagent (Thermofisher Scientific, CR). The active component of this assay – resazurin is reduced to a fluorescently active compound resorufin only in viable/metabolically active cells, thus the fluorescence intensity directly correlates with the number of viable cells. The coverslips were incubated with AB reagent for 4 h at 37 °C and the fluorescence was detected in a multi-well plate reader Syner Neo (Bio-Tek, CR) using excitation at 570 nm and emission at 600 nm. The acquired fluorescence of the samples was re-calculated using a calibration curve in order to obtain the absolute cell number present on the samples. For the calibration curve, six wells in duplicates were seeded with 5000–50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells day before and the fluorescence of AB metabolized component was measured. Then the cells were washed with PBS and incubated with 0.2 ml of trypsin solution (0.05% trypsin and 0.5 mM EDTA in PBS) at 37 °C for 5 min. The trypsin was deactivated by adding 0.2 ml of media. The solution was aspirated from each well and the cell pellet was re-suspended in 100 μl of PBS. The Bruker's chamber was used to determine the number of cells. The 0.2% Trypan blue – lethal dye was used to assess number of dead cells and correlate only live cells with appropriate fluorescence intensity.32 The calibration curve from the number of live cells growing on the well plates and their fluorescence were used for the determination of the number of attached and growing cells on each coverslip according to their fluorescence intensity. Each surface modification was tested in triplicate.
000 cells day before and the fluorescence of AB metabolized component was measured. Then the cells were washed with PBS and incubated with 0.2 ml of trypsin solution (0.05% trypsin and 0.5 mM EDTA in PBS) at 37 °C for 5 min. The trypsin was deactivated by adding 0.2 ml of media. The solution was aspirated from each well and the cell pellet was re-suspended in 100 μl of PBS. The Bruker's chamber was used to determine the number of cells. The 0.2% Trypan blue – lethal dye was used to assess number of dead cells and correlate only live cells with appropriate fluorescence intensity.32 The calibration curve from the number of live cells growing on the well plates and their fluorescence were used for the determination of the number of attached and growing cells on each coverslip according to their fluorescence intensity. Each surface modification was tested in triplicate.
Results and discussion
Characterization of polymers and their colloid solutions
The A–B diblock copolymers composed of the semi-crystalline polylactide block (PLLA) with a matching block length (theor. 7 kDa) and the semi-crystalline methoxy-poly(ethylene oxide) block (PEO) with a block length of 5, 10, and 15 kDa were synthesized by a ring-opening polymerization and characterized by 1H NRM spectroscopy and SEC. The copolymer composition was calculated from the proton spectrum (see Fig. S1 in the ESI† for 1H NMR spectra) from the ratio of integrated peaks of methylene protons of the PEO unit (3.54 ppm) and that of the PLLA-methine proton (5.2 ppm). The increasing molecular weight of the PEO block is clearly demonstrated by the incremental ratio of the PEO unit at 3.54 ppm to the PLLA one at 5.2 ppm. The molecular parameters of the prepared PLLA-b-PEO copolymers are listed in Table 1.Amphiphilic block copolymers such as PLLA-b-PEO can form in aqueous solutions or selective solvent self-assemblies, particularly core–shell nanoparticles.16,33 The character of self-assemblies of a wide range of PEO-based amphiphilic block copolymers was investigated in a comparative study by Dionzu et al.34 The hydrophilic/hydrophobic fraction was shown to be a significant factor for prediction of the morphology of emerging nano-objects, which are not usually in equilibrium, but kinetically frozen. However, besides molecular parameters, the threshold between micelles and vesicles also depends on details such as (co)solvent system and preparation method. In the mentioned study, the micelle formation of PLA-b-PEO copolymers was observed for the copolymers with higher hydrophilic weight fractions fPEO, particularly for fPEO > 0.4.
In the present study, we used the mixed solvent of acetone and methanol (1/9 v/v). Methanol served as a coagulant for the PLLA phase,35 thus forming the core from the collapsed chains of the PLLA blocks, and as a good solvent for the PEO blocks generating the particle shell. The hydrophilic weight fraction fPEO reached the values 0.42, 0.56, and 0.68 for the 7/5, 7/10, and 7/15 PLLA-b-PEO copolymers indicating formation of micelle morphology according to Dionzu's work.34 The formation of such self-assemblies is shown in Fig. 1A. For all tested copolymers, opalescent solutions, i.e. suspensions were obtained and further characterized by DLS. The hydrodynamic radii RH of the formed nanoparticles of the 7/5, 7/10, and 7/15 PLLA-b-PEO copolymers are given in Table 1. The results clearly show that the size of the PLLA-b-PEO nanoparticles increases with increasing PEO block length, which is in agreement with the theory of “hairy” micelles.36 Besides, the nanoparticles become more regular and uniform with increasing content of the hydrophilic PEO block as indicated by narrowing of the size distribution given by PdI (Table 1).
The mixed solvent composed of acetone and methanol afforded formation of the PLLA-b-PEO particles with an approximately tenfold higher diameter when compared with nanoparticles prepared from a dioxane–water solvent system,16 or prepared by dialyzing the PEG-PLA block copolymer in dimethylacetamide against deionized water.33 We prepared nanoparticles of 7/5 PLLA-b-PEO with diameters of 170 nm up to 230 nm for 7/15 PLLA-b-PEO.
Characteristics of PLLA-b-PEO layers formed on the PLLA substrate
The model surfaces of the PLLA-b-PEO copolymers on the poly(lactide) support were prepared as thin layers spin-cast on the PLLA film. Here the PLLA film mimics the surface of a real biomaterial. A simple sample drying at laboratory temperature led to the copolymer coatings becoming unstable under aqueous conditions, as was observed as changes in the surface wettability when the surfaces were incubated in water for several days (data not presented). Therefore to anchor the copolymer layers, the surfaces were exposed to a temperature of 50 °C under vacuum followed by annealing in water at 40 °C and finally by quenching in freezing water. The temperatures of 40 and 50 °C were selected as they are close to the glass transition temperature of PLLA and to the melting temperature of PEO. It was expected that such a layer treatment could enhance the stability of the copolymer layer on the support due to the enhanced phase separation of the PEO and PLLA blocks and due to the entanglement of the polylactide chains from the copolymer with the polylactide chains of the PLLA support layer. Furthermore, water annealing was used to enhance additional partial reorientation of the PEO chains towards the water environment to form the upper film layer enriched with the hydrophilic PEO. Indeed, the thermal treatments resulted in the copolymer surfaces being stable in aqueous conditions at 37 °C at least for 1 week as verified by CA measurements and ellipsometry analysis (see Table S1 in the ESI†). The surfaces prepared from the copolymers with amorphous poly(DL-lactide) blocks showed poor stability under physiological conditions (PBS, 37 °C). In contrast, PLLA-b-PEO copolymers with semi-crystalline poly(L-lactide) blocks together with a stabilization procedure at increased temperature provided hydrophilic surfaces that are stable under cell culture conditions. Table 2 reports the average ellipsometric thickness and calculated surface-related parameters of the PLLA-b-PEO block copolymer layers on the PLLA substrate. The spin coating conditions were adjusted so the copolymer layer thickness (h) would reach about 20 nm irrespective of the molecular weight of the PEO block. The approximated PLLA-b-PEO chain density (σ) on PLLA decreased from 0.9 to 0.6 chain nm−2 with increasing the molecular weight of the PEO blocks from 5 to 15 kDa. The decrease in σ in turn caused an increase in distance (D) between the polymer chains from 1.1 to 1.4 nm. In all cases the PEO blocks showed a strong inter-chain overlapping, i.e.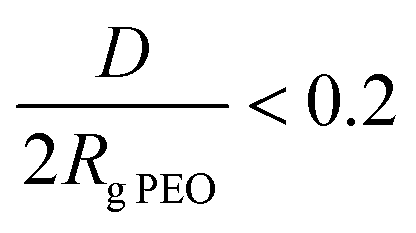 implying that the PEO chains will adopt a brush structure in contact with water. The dense anchoring of the copolymer chains on the PLLA substrate is further corroborated by the decrease in advancing CA (Table 2) from 83 ± 1° for the pristine PLLA film to 55 ± 2°, 36 ± 3° and 34 ± 3° for the 7/5, 7/10 and 7/15 PLLA-b-PEO layers, respectively. Furthermore, the presence of PEO in the top layer of the film is documented by a significant diminution of the receding CA when compared to the PLLA surface. We attributed this observation to the swelling and penetration of water into the PEO chains resulting in their mobility and reorientation. The receding contact angle slightly decreases with the increasing length of the PEO block. According to the contact angle theory, these observations indicate the presence of high energy, i.e. a low-angle PEO phase in the top layer.37
implying that the PEO chains will adopt a brush structure in contact with water. The dense anchoring of the copolymer chains on the PLLA substrate is further corroborated by the decrease in advancing CA (Table 2) from 83 ± 1° for the pristine PLLA film to 55 ± 2°, 36 ± 3° and 34 ± 3° for the 7/5, 7/10 and 7/15 PLLA-b-PEO layers, respectively. Furthermore, the presence of PEO in the top layer of the film is documented by a significant diminution of the receding CA when compared to the PLLA surface. We attributed this observation to the swelling and penetration of water into the PEO chains resulting in their mobility and reorientation. The receding contact angle slightly decreases with the increasing length of the PEO block. According to the contact angle theory, these observations indicate the presence of high energy, i.e. a low-angle PEO phase in the top layer.37
| Layer | h [nm] | σ [chain per nm2] | D [nm] | D/2Rg PEO | Θ A [°] | Θ R [°] |
|---|---|---|---|---|---|---|
| PDA | 4.0 ± 0.4 | — | — | — | 62 ± 4 | 18 ± 2 |
| PLA | 25.5 ± 3.2 | — | — | — | 83 ± 1 | 65 ± 1 |
| 7/5 | 19.7 ± 0.8 | 0.9 | 1.1 | 0.19 | 55 ± 2 | 23 ± 3 |
| 7/10 | 20.0 ± 2.1 | 0.7 | 1.3 | 0.15 | 36 ± 3 | 10 ± 2 |
| 7/15 | 19.6 ± 1.0 | 0.6 | 1.4 | 0.13 | 34 ± 3 | 8 ± 2 |
The IRRAS spectra of the prepared copolymer films (Table 1) and the homopolymer PLLA and PEO films are presented in Fig. 2. The representative spectrum of the PLLA-containing films is dominated by bands at 2997, 2947, and 2870 cm−1 assigned to the asymmetric CH3, symmetric CH3, and CH stretching modes, respectively. The strongest observed band at 1769 cm−1 arises from the CO stretching modes. The band at 1456 cm−1 is assigned to the symmetric deformation of CH3, while the bands at 1387 and 1361 cm−1 are assigned to the CH deformation and asymmetric deformation of CH3. The bands at 1137 and 1045 cm−1 can be assigned to asymmetric rocking of CH3 and skeletal C–CH3 stretching. The asymmetric C–O stretching modes of the ester group appear as strong bands at 1218 and 1186 cm−1, while the symmetric C–O mode gives rise to the band at 1090 cm−1. The C–COO stretching vibrations give rise to the weak band at 873 cm−1.38
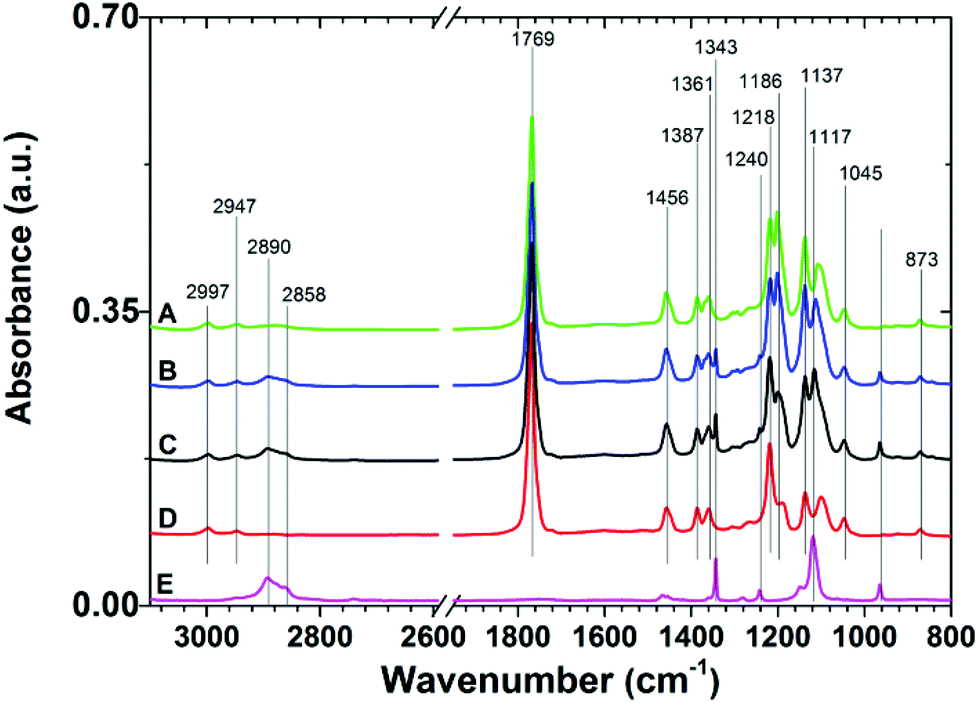 | ||
| Fig. 2 Representative IRRAS spectra of 7/5 (A), 7/10 (B), and 7/15 (C) PLLA-b-PEO/PLLA thin polymer films on a PDA adhesion layer. Spectra of PLLA (D) and PEO (E) are presented for comparison. | ||
The spectra of the PLLA-b-PEO copolymer films, in addition to the contributions characteristic for the PLLA block, show spectral features of PEO. In the C–H stretching region the presence of PEO gives rise to bands at 2890 and 2858 cm−1 from the stretching modes of CH2 of the ethylene oxide units. Most of the expected bands (arising from CH2 rocking (964 cm−1), twisting (1281 and 1242 cm−1), wagging (1360 cm−1), and scissoring (1467 and 1455 cm−1) modes) strongly overlap with the contributions from the PLLA block. Importantly, we also observed the distinctive bands of the C–O–C stretching mode at 1343 and 1117 cm−1 in the fingerprint region. These contributions have been assigned to the PEO chains in helical conformation with a predominant perpendicular orientation with respect to the surface as in the case of the dense PEO brushes end-tethered directly to gold39 or to a PDA anchor layer.7,25 Thus the presence of these bands proves the brush structure of the PEO blocks in the top layer of the PLLA-b-PEO films in our study. Notably, the spectral contributions of PEO rise with increasing molecular weight of the PEO block.
The presence of the PLLA-b-PEO copolymers on the PLLA surfaces was further corroborated by the XPS analysis (Fig. 3 and S3 in the ESI†). The high resolution C 1s XPS spectrum of pure PLLA films is characterized by almost equal contributions of the C–C, C–O and O–CO moieties at 285.0, 286.7 and 289.0 eV, respectively (Fig. 3D). The high resolution O 1s XPS spectrum (see Fig. S3D in the ESI†) of PLLA shows two contributions from the O–C and OC moieties at 532.3 and 533.8 eV. Expectedly, the C 1s and O 1s spectra of pure PEO films show prevailing C–O–C peaks at 286.4 eV and at 532.5 eV, respectively. The immobilization of the PLLA-b-PEO chains on the PLLA substrate leads to clear changes within the C 1s and O 1s high resolution spectra. While the C–O–C contributions arising from EO monomer units rise with the increasing molecular weight of the PEO block, we observe a concomitant drop of the characteristic C–C and O–CO contributions arising from the lactide monomer units (see Fig. 3A–C). Similar decreasing trends are observed in the O 1s high resolution spectra for the OC (Fig. S3 in ESI†) which are characteristic for the lactide units of the PLLA block. Furthermore, the (O–CO)PLLA/(C–O)PEO and (OC)PLLA/(O–C)PEO ratios were calculated on the basis of the high resolution C 1s and O 1s XPS spectra of the copolymer films (see Table S2 in the ESI†). The agreement between the experimental and theoretical ratios underlines the successful modification of the PLLA surfaces with compact films of the PLLA-b-PEO block copolymers.
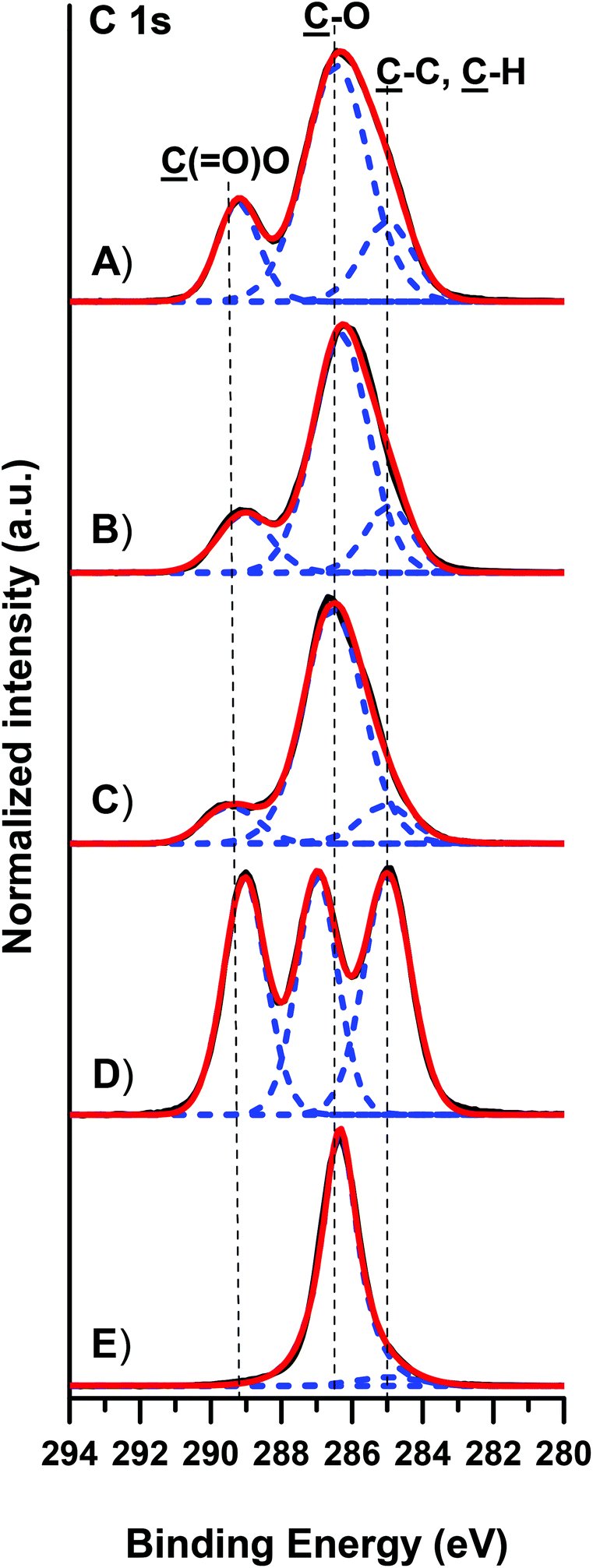 | ||
| Fig. 3 Representative high resolution C 1s XPS spectra of 7/5 (A), 7/10 (B), and 7/15 (C) PLLA-b-PEO/PLLA thin polymer films on a PDA adhesion layer. Spectra of PLLA (D) and PEO (E) are presented for comparison. | ||
The topography of the (co)polymer surfaces was investigated using AFM both in air and in PBS. The micrographs taken for the plain PLLA substrate are depicted in Fig. S2 in the ESI† and those taken for the PLLA-b-PEO surfaces are shown in Fig. 4. As is clearly seen from Fig. 4B and F, the topography of the surfaces composed of the copolymers with a longer PEO block, i.e. 7/15 and 7/10 PLLA-b-PEO layers, exhibited in air (dry state) an evident lamellar structure. Although the overall topography of both surfaces is similar, the average diameter of the observed lamellas decreases with the decreasing molecular weight of the PEO block from 26 nm in the case of the 7/15 surface to 18 nm for the 7/10 surface. However, as is evident from the 1 × 1 μm detailed scan images (Fig. 4B, F, and J), the distinct lamellar structure disappeared with the further decrease in the PEO block length to 5 kDa (Fig. 4J). The micrograph shows that the 7/5 copolymer surface is formed from unorganized, partly round- and ellipsoid-shaped hills with a hardly detected lamellar structure of the separated PEO blocks. Our observation thus provides evidence that the length of the PEO block plays a driving role in the formation of the surface structure. This observation correlates with our previous study on PDLLA-b-PEO surfaces with amorphous polylactide blocks14 where we observed a lamellar structure on the surface with the 15 kDa PEO block while the surface of the copolymer with the 5 kDa PEO block exhibited round shaped features. Based on the obtained results, we can attribute the lamellar structure to the ability of PEO to crystalize during the film casting and the film drying before the AFM analysis. Furthermore, the root mean squared surface roughness (RRMS) increased from 4.6 to 5.7 and to 9.4 nm with the decreasing length of the PEO blocks from 15 to 5 kDa. The observed decrease in RRMS with increasing PEO length is in agreement with the observations published for the topography of magnetic poly(glycidyl methacrylate) microspheres grafted with PEO of a different length.40
 | ||
| Fig. 4 AFM topography images of PLLA-b-PEO copolymer surfaces 7/15 (A, B, C, D), 7/10 (E, F, G, H), and 7/5 (I, J, K, L) taken in air (A, B, E, F, I, J) and in PBS (C, D, G, H, K, L). | ||
The images taken in PBS are presented in Fig. 4C, D, G, H, K, and L. It is clearly evident from the 5 × 5 μm scans of a larger area (Fig. 4C, G and K) that the 7/5 surface exhibits a noticeably more grain-type pattern than the 7/10 and 7/15 surfaces with longer PEO blocks; and the surface roughness increases from 7.5 to 10.0 and 16.2 nm with the decreasing length of the PEO chains. These differences in topography correlate with the observations on the dry surfaces. Although differences in the surface structure in the 1 × 1 μm detailed scan images in PBS (Fig. 4D, H and 4L) are not so obvious, we consider the lamellar structure on the 7/15 and 7/10 surfaces to be still distinguishable, however with a lower definition in comparison with the morphology in the dry state. We assume that the granular-type topography observed in Fig. 4L represents simply the swollen structures observed in Fig. 4J. The granular character could reflect the lower molar weight of the PEO block in comparison with the lamellar structures observed for copolymers with 10 kDa and 15 kDa PEO blocks. The difference in the topography of swollen and dry surfaces can result from the drying process which can induce faint re-orientation of the PEO chains. Indeed, the roughness of the surfaces swollen in PBS is one and a half times higher than that of the surfaces in the dry state. The lamellas appeared to be more swollen in PBS which indicates that the present PEO chains are hydrated and expanded when in contact with water. Importantly, as is clearly seen from the presented AFM micrographs, the initially observed structure in air (e.g.Fig. 4F) is preserved after the re-immersion in PBS (e.g.Fig. 4H) with the only difference of a significant swelling due to water uptake.
The comparison of our AFM observation with other studies is limited as the surface topography is influenced by the molecular parameters of the used copolymers, by conditions of deposition including concentration and a (co-)solvent system and annealing as well as by the character of the supporting surface.41 Furthermore, in the case of amphiphilic block copolymers with the potential ability to crystallize, the hydrophilic to hydrophobic block ratio and thus the phase separation plays an important role in the formation of surfaces with the ordered structures.42 Besides, model surfaces, such as mica, glass or Si wafers are mostly used for the investigation of the surface morphology of PLA-b-PEO copolymers.
For example, Huang et al. studied the morphology of isothermally crystallized thin films of asymmetric PLLA-b-PEO copolymers.43 The observed dendritic superstructures were attributed to the crystallization of PLLA blocks and to microphase separation between PLLA and PEO blocks. However, comparison with our study is unfeasible as the PLLA to PEO block length ratios (16/5 and 30/5) and the conditions of the surface preparation were different from those used in our study.43
The deposition of a PLA-b-PEO copolymer with comparable parameters and under similar conditions was studied by Emoto et al.33 The deposition of reactive micelles with a cross-linked core of PDLLA-b-PEO 3.5/4.5 onto the various substrates resulted in the presence of lumps, as investigated by AFM both in air and in water, and it indicated the deposition of whole micelles. However, the distinct spherical micelle structure was not observed when the micelles with a non-cross-linked core were deposited on the wafers. In this case the topography exhibited regular hill features indicating interaction of the uncovered micelle core with the surface according to the model proposed by Ligoure.22 Furthermore, Fujiwara et al. studied the surface deposition of PLLA-b-PEO 4.5/5 copolymers from water solutions of different concentrations and the effect of thermal annealing on topography.42 The study has shown that the PLLA-b-PEO micelles cannot preserve their shape after deposition on the surface from diluted solutions. On the other hand, at higher PLLA-b-PEO concentrations (from 0.2 wt%), spherical hills were observed on the surface. Moreover, annealing at 60 °C changed the surface topography to a sphere-band structure.
Based on Popelka's study,16 where the deposition of whole PLA-b-PEO micelles with non-cross-linked cores was observed, and considering the copolymer concentration, solvent system, and Ligoure model,22 we assume that in our system, when the PLLA core is in a frozen state in a selective solvent, the PLLA-b-PEO copolymers can be deposited on the PLLA substrate as nanoparticles. The PLLA-b-PEO nanoparticles can relax upon contact and the core will interact with the PLLA substrate while PEO will be exposed to the outer environment providing hydrophilic surface.
The specific surface structures observed by AFM are provided by the difference in the PLLA/PEO block ratio and the PEO length itself, hence the PEO ability to crystalize and by stabilization of the deposited copolymer films at 50 °C and water annealing at 40 °C.
Considering our results from IRRAS, XPS and spectroscopic ellipsometry analysis together with contact angle values and the changes in topography, we can conclude that in the case of the polymer surfaces prepared by the deposition of colloid solutions of the PLLA-b-PEO copolymers, the PEO chains adopt the brush structure and are preferably exposed at the top layer of the stack. When exposed to an aqueous environment the PEO chains swell and further re-orient and reorganize.
Protein adsorption and cell adhesion
The protein-resistance properties of the tested surfaces were determined using SPR spectroscopy. For this purpose, the PLLA-b-PEO (7/15; 7/10; 7/5) layers and the reference surfaces, i.e. the pure PLLA layer and a gold SPR chip were exposed to a single plasma protein solution (human serum albumin (HSA) or fibrinogen (Fbg)) as well as to the cell cultivation media (Minimum Essential Medium Eagle (MEME) supplemented with 10% FBS and fetal bovine serum (FBS, 100%)) for 0.5 h. The protein adsorption was observed in situ as a shift in the resonance wavelength. A typical SPR record is shown in Fig. S4 in the ESI.† The mass of the adsorbed proteins onto the surfaces calculated according to Kumorek et al.44 is presented in Fig. 5.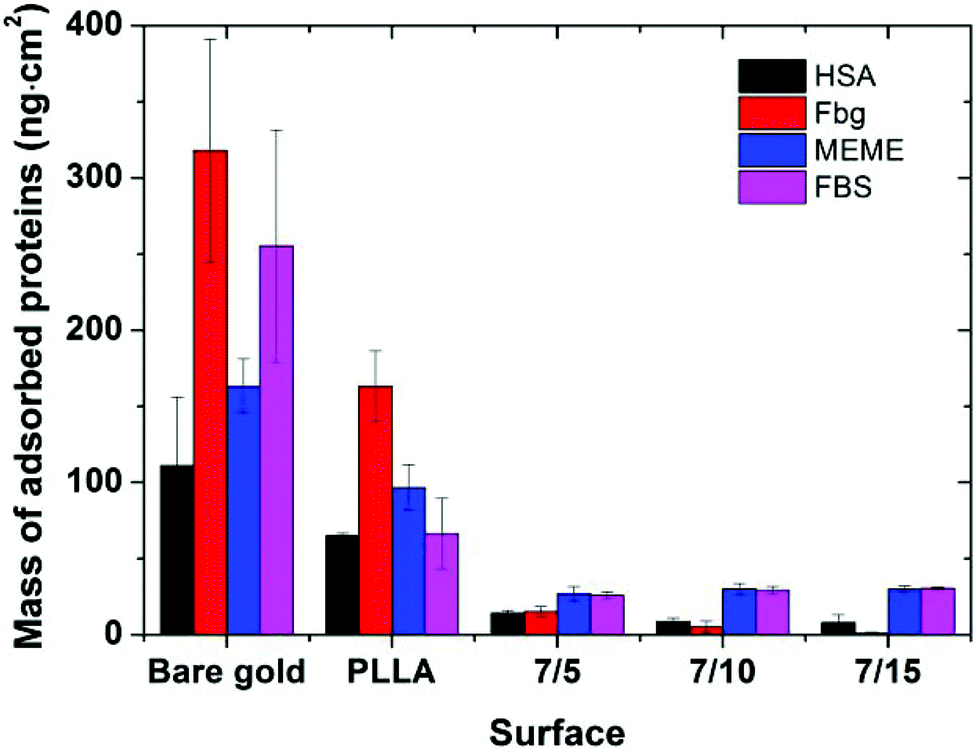 | ||
| Fig. 5 Protein adsorption on PLLA and PLLA-b-PEO (7/5; 7/10; 7/15) layers bound to PDA followed by SPR. The surfaces were exposed to HSA (5 mg ml−1 in PBS), Fbg (1 mg ml−1 in PBS), MEME (supplemented with 10% FBS), and FBS (100%). A bare gold chip was used as a reference. Data are presented as mean ± standard deviation. | ||
When compared with the gold surface, a partially reduced protein adsorption was observed on the PLLA film. However, the surface coverage by proteins is still high and such a surface cannot be considered as a suitable background for the creation of smart biomimetic functional surfaces capable of eliciting accessible integrin binding ligands.
Considering the copolymer surfaces, all PLLA-b-PEO films revealed good protein-resistance properties against single plasma proteins, i.e. Fbg and HSA. The adsorption of Fbg, often referred as the “sticky protein”, to the 7/5 PLLA-b-PEO surface was determined to be only 15 ± 3 ng cm−2, i.e. only 9% of the Fbg layer formed on the pure PLLA surface (163 ng cm−2). Remarkably, the Fbg adsorption on the 7/15 surface with the longest PEO block was close to the detection limits of the measurement, which indicates a complete prevention of the non-specific adsorption of Fbg. Similarly, the adsorption of HSA, the most abundant protein in blood plasma, dropped from 65 ng cm−2 on PLLA to 14 ± 2 ng cm−2 on the 7/5 PLLA-b-PEO surface, and further down to 8 ± 5 ng cm−2 with the increasing molecular weight of the PEO block of 15 kDa. The results revealed that the adsorption of both proteins decreases with the increasing length of the PEO blocks, i.e. with the increasing brush structure of the PEO chains indicated by the drop of the overlapping parameter 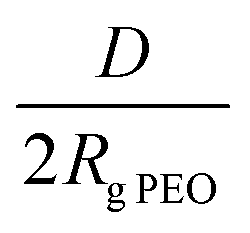 from 0.19 to 0.13 (Table 2).
from 0.19 to 0.13 (Table 2).
Interestingly, Otsuka et al. observed the lowest adsorption of albumin onto the PLLA-b-PEO surfaces with shorter PEO segments, particularly approx. 3 kDa and not onto the surface with the longest PEO chain tested (9 kDa).15 This discrepancy with our findings can be caused by a different process of sample preparation, leading to differences in the characteristics of the prepared surfaces including the PEO grafting densities. In the mentioned study, the copolymer surfaces were prepared by spin-casting from toluene solution of PLLA-b-PEO (20 mg ml−1), i.e. from the good solvent for both blocks. The samples were dried under vacuum at room temperature. Such a procedure resulted in moderate-hydrophilic surfaces. Although the PEO grafting density, the state of the grafted chains and the chain overlapping parameters were not explicitly determined in the referred work, the authors assumed that in their system the PEO blocks reached the maximum of the chain density on the surface with shorter PEO chains.15 However, more importantly, our procedure of PLLA-b-PEO surface preparation provided copolymer layers with markedly lower adsorption of albumin of more than one order of magnitude regardless of the PEO block lengths compared to Otsuka et al.15
When the PLLA-b-PEO surfaces were brought into contact with the multicomponent biological solutions MEME and FBS, the notorious fouling observed on the gold and PLLA reference surfaces was also significantly suppressed. The mass of the adsorbed protein components decreased to less than 30 ng cm−2 regardless of the length of the PEO chain. Notably, such a low level of non-specifically adsorbed protein deposits approaches the levels reported for ultra-low and low-fouling PEO polymer brush systems prepared by grafting from a reactive melt7,25,45 and polymer brushes synthesized by surface initiated controlled atom transfer radical polymerizations.30
It is evident that the PEO blocks in aqueous solutions can be swollen and expanded and play an important role in significant reduction of fouling from complex protein solutions tested on the PLLA-b-PEO surfaces. Additionally, in the case of single protein solutions, the SPR data showed a noticeable effect of the PEO block length on the anti-fouling surface properties.
The observations from the cell culture experiments with the MG63 cell line are in a good agreement with the trends obtained from the protein adsorption experiments. The further concept of such a surface modification functionalized with biomimetic motifs is mainly proposed for the interaction with bone cells. Consequently, the MG63 cell line has been used as a representative model of bone cells. The HUVEC cell line has been used to confirm the general properties of the prepared surfaces. PLLA has a rather hydrophobic surface which tends to provoke higher protein adsorption due to strong hydrophobic interactions. Consequently, MG63 attached to the PLLA surface within 24 hours as manifested by the cell spreading with a typical morphology (see Fig. 6A). In contrast, all three PLLA-b-PEO surfaces, which exhibited a hydrophilic character (Table 1) providing the repulsive solvation forces4 and thus significantly suppressed adsorption of single proteins as well as complex proteins from MEME, did not enable spreading of MG63 within 24 hours after the cell seeding as the PLLA surface. The cell morphology remained roundish (Fig. 6B–D) indicating poor cell attachment to the substrates and the cells’ inability to grow. A similar pattern of attachment was observed with HUVEC cells (Fig. S5†).
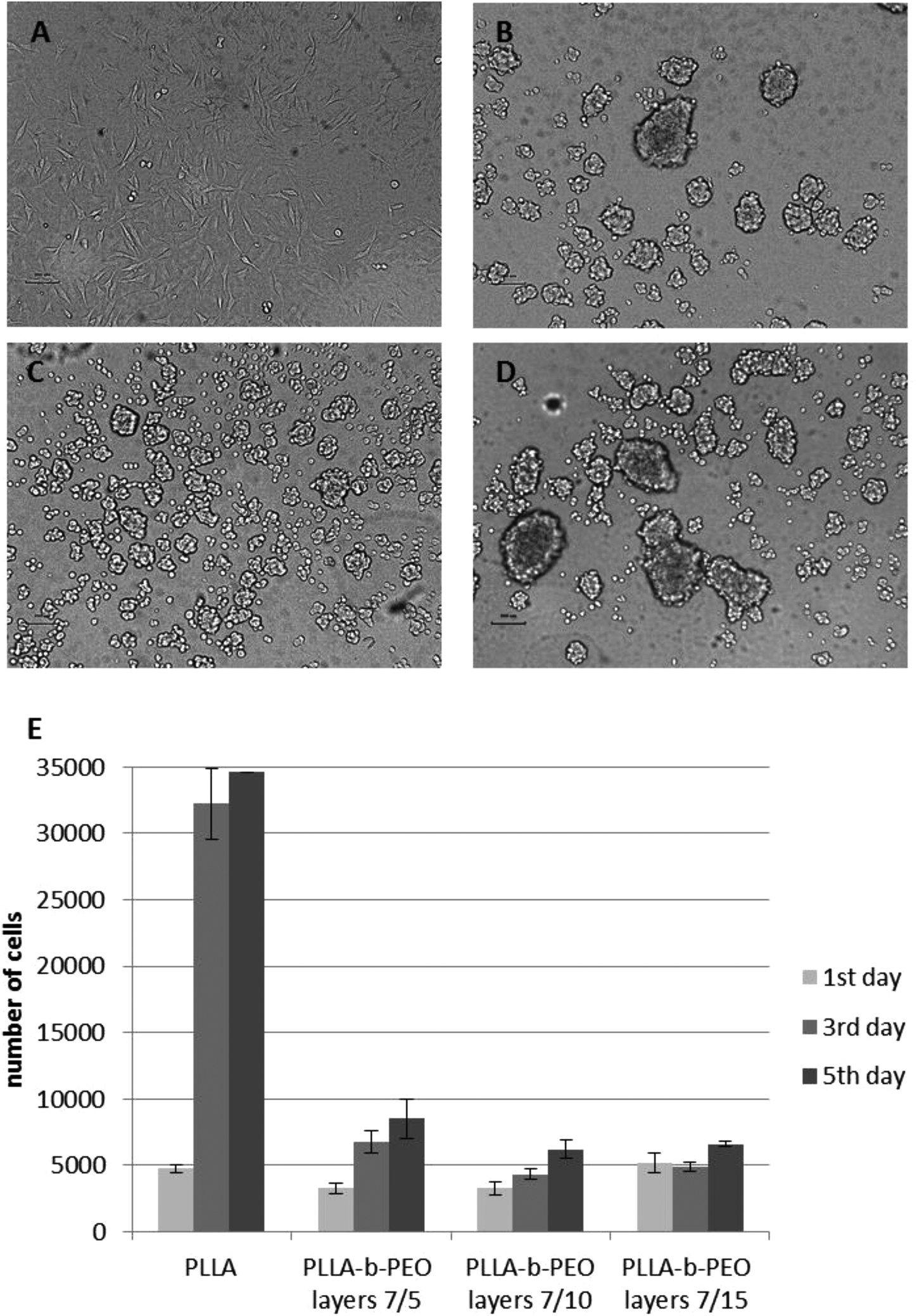 | ||
| Fig. 6 Evaluation of initial attachment and proliferation MG63 cells to the prepared surfaces. Light microscopy of the attachment of MG63 to the plain PLLA surface (A) and PLLA-b-PEO layers 7/5 (B), 7/10 (C), and 7/15 (D) after 24 hours since cell seeding. Scale bar: 200 μm. (D) Calculation of cell numbers after 1, 3 and 5 days of growth on the surfaces using cell viability assay. | ||
MG63 has been also characterized as the cell number of attached cells after 24 hours and the cell number of growing cells at day 3 and 5 (Fig. 6E). The number of cells 24 hours after the seeding was not significantly different among the surfaces even though the morphology of the cells was significantly different. After 24 hours on PLLA-b-PEO surfaces, the cells were spread over a relatively small area and weakly adherent; and hence, with inappropriate stimuli for further growth. This phenomenon has been confirmed by a significant difference in the cell number at day 3 and 5 among PLLA and PLLA-b-PEO surfaces. On the PLLA the cell proliferation has reached more than 30000 cells per slide. In contrast, on the PLLA-b-PEO surfaces the cells did not proliferate properly, keeping the cell number between 4000–8000 cells per slide and remaining within a round morphology.
Based on the cell culture experiments together with the SPR results, we can assume that the modification of the PLLA surface with the PLLA-b-PEO colloids provides the surfaces with abundantly reduced protein adsorption sufficient for the prevention of cell adhesion within the range of the PEO molecular weight from 5 kDa to 15 kDa. Hence, as such, when a portion of the PEO chains is modified with a biomimetic component, the colloids can be used for tailored nanostructured and nano-patterned surfaces with adjustable distance and density of biomimetic peptides, which would not be overlaid by the mass of the proteins adsorbed from the media and will be available for interaction with cell receptors.
Apart from low fouling properties of the present PLLA-b-PEO coating we take into consideration the feasibility of this coating to provide patterned surfaces in a controlled manner. The proposed procedure afforded formation of the PLLA-b-PEO particles with the diameter about 200 nm. We expect that the size of the copolymer nanoparticles in hundreds of nanometers will be more suitable in the preparation of functionalized surfaces, where the intended patches of biomimetic motifs can be prepared and controlled in proportions related to the dimension of the cells and the cell receptors. Such a surface modification can be difficult to achieve by the deposition of other PEO-based nanoparticle systems with a lower diameter of several or tens of nanometers such as PEO-PLA micelles16,33,42 or pluronic-based nanoparticles, although they exhibit low-fouling properties.46,47
Although in this study we focused only on PLLA-b-PEO copolymers modifying PLLA surfaces, the approach can be applied also for analogous combinations of amphiphilic block copolymers composed of PEO and other linear polyester blocks, e.g. polycaprolactone, or lactide–glycolide copolymers, when modifying corresponding polyester surfaces. The mentioned polyester-b-PEO can readily be prepared through the analogous chemistry to the PLA-b-PEO copolymers. Therefore, we believe that the colloidal deposition approach could be similarly applied to polyester-based biomaterials, which represent an important class of biomedical polymers.
Conclusions
Polylactide surfaces can be effectively modified by deposition of amphiphilic PLLA-b-PEO copolymers in the form of colloids from selective solvents resulting in hydrophilic anti-fouling surfaces. The surface morphology, efficiency of the modification, and the anti-fouling character depends on the molecular parameters of the used copolymers. The PLLA-b-PEO self-assemblies formed in selective solvents were deposited on the PLLA surface as stable nanoparticles with a kinetically frozen PLLA core; the average size of the nanoparticles increased with increasing PEO block length. Upon deposition, the nanoparticles can relax providing tight contact between the PLLA core and the PLLA surface, and hence, the PEO corona adopted the brush structure in the top layer. The brush structure was proved by spectroscopic ellipsometry and IRRAS analysis for all tested copolymer films. Furthermore, the different topography of the dry copolymer surfaces was attributed to the ability of the PEO blocks to crystallize. The slight difference in the topography observed for the wetted surfaces resulted from swelling of the top PEO layer; hence, hydrated PEO brushes provided repulsive forces to proteins.The most efficient surface modification was achieved with the PLLA-b-PEO copolymer with a PEO block of 15 kDa, providing highly hydrophilic surfaces with a significantly suppressed adsorption of albumin and fibrinogen. Moreover, although the copolymer with the short PEO block, i.e. 5 kDa exhibited only a moderate hydrophilic character, it significantly repelled protein and cell adhesion as compared to the pure PLLA surface. The anti-fouling properties of the PLLA-b-PEO layers in the cell cultivation media, i.e. MEME and FBS, regardless of the length of the PEO block, approximated the levels obtained on anti-fouling PEO brushes prepared by grafting techniques. Therefore, all tested PLLA-b-PEO copolymers can be considered as a protein resistant enough to provide a non-cell-adhesive polymer background for further modification with biomimetic compounds of smart surfaces in the area of tissue engineering.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
The support by the Czech Science Foundation (GACR, Grant No. 15-09368Y), by Czech health research council, Ministry of Health of the Czech Republic (No.16-28254A) and the Ministry of Education, Youth and Sports of CR within the National Sustainability Program II (Project BIOCEV-FAR LQ1604), and by the project “BIOCEV” (CZ.1.05/1.1.00/02.0109) from the European Regional Development Fund is acknowledged.References
- O. Pop-Georgievski, C. Rodriguez-Emmenegger, A. Santos Pereira, V. Proks, E. Brynda and F. Rypacek, Biomimetic non-fouling surfaces: extending the concepts, J. Mater. Chem. B, 2013, 1, 2859–2867 RSC.
- Biomimetic biomaterials: Structure and Applications, ed. A. J. Ruys, Woodhead Publishing, 1st edn, 2013 Search PubMed.
- G. M. Harbers, K. Emoto, C. Greef, S. W. Metzger, H. N. Woodward, J. J. Mascali, D. W. Grainger and M. J. Lochhead, A functionalized poly(ethylene glycol)-based bioassay surface chemistry that facilitates bio-immobilization and inhibits non-specific protein, bacterial, and mammalian cell adhesion, Chem. Mater., 2007, 19, 4405–4414 CrossRef CAS PubMed.
- H. Chen, L. Yuan, W. Song, Z. Wu and D. Li, Biocompatible polymer materials: Role of protein-surface interactions, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS.
- X. Ma, Y. Su, Q. Sun, Y. Wang and Z. Jiang, Enhancing the antifouling property of polyethersulfone ultrafiltration membranes through surface adsorption-crosslinking of poly(vinyl alcohol), J. Membr. Sci., 2007, 300, 71–78 CrossRef CAS.
- J. H. Lee, H. B. Lee and J. D. Andrade, Blood compatibility of polyethylene oxide surfaces, Prog. Polym. Sci., 1995, 20, 1043–1079 CrossRef CAS.
- O. Pop-Georgievski, D. Verreault, M.-O. Diesner, V. Proks, S. Heissler, F. Rypáček and P. Koelsch, Nonfouling Poly(ethylene oxide) Layers End-Tethered to Polydopamine, Langmuir, 2012, 28, 14273–14283 CrossRef CAS PubMed.
- I. Szleifer, Protein adsorption on surfaces with grafted polymers: a theoretical approach, Biophys. J., 1997, 72, 595–612 CrossRef CAS PubMed.
- M. A. Carignano and I. Szleifer, Prevention of protein adsorption by flexible and rigid chain molecules, Colloids Surf., B, 2000, 18, 169–182 CrossRef CAS PubMed.
- A. Halperin, Polymer brushes that resist adsorption of model proteins: design parameters, Langmuir, 1999, 15, 2525–2533 CrossRef CAS.
- D. Leckband, S. Sheth and A. Halperin, Grafted poly(ethylene oxide) brushes as nonfouling surface coatings, J. Biomater. Sci., Polym. Ed., 1999, 10, 1125–1147 CrossRef CAS PubMed.
- E. P. K. Currie, W. Norde and M. A. Cohen Stuart, Tethered polymer chains: surface chemistry and their impact on colloidal and surface properties, Adv. Colloid Interface Sci., 2003, 100–102, 205–265 CrossRef CAS PubMed.
- R. A. Quirk, M. C. Davies, S. J. B. Tendler and K. M. Shakesheff, Surface engineering of poly(lactic acid) by entrapment of modifying species, Macromolecules, 2000, 33, 258–260 CrossRef CAS.
- E. Tresohlava, S. Popelka, L. Machová and F. Rypacek, Modification of polylactide surfaces with lactide-ethylene oxide functional block copolymers: accessibility of functional groups, Biomacromolecules, 2010, 11, 68–75 CrossRef CAS PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Surface characterization of functionalized polylactide through the coating with heterobifunctional poly(ethylene glycol)/polylactide block copolymers, Biomacromolecules, 2000, 1, 39–48 CrossRef CAS PubMed.
- S. Popelka, L. Machova, F. Rypacek, M. Spirkova, M. Stepanek, P. Matejicek and K. Prochazka, Dynamics of chain exchange between self-assembled diblock copolymer micelles of poly(ethylene oxide)-block-polylactide studied by direct nonradiative excitation energy transfer, Collect. Czech Chem. Commun., 2005, 70, 1811–1828 CrossRef CAS.
- P. Knotek, E. Chanova and F. Rypacek, AFM imaging and analysis of local mechanical properties for detection of surface pattern of functional group, Mater. Sci. Eng., C, 2013, 33, 1963–1968 CrossRef CAS PubMed.
- M. Arnold, E. A. Cavalcanti-Adam, R. Glass, J. Blummel, W. Eck, M. Kantlehner, H. Kessler and J. P. Spatz, Activation of integrin function by nanopatterned adhesive interfaces, ChemPhysChem, 2004, 5, 383–388 CrossRef CAS PubMed.
- E. A. Cavalcanti-Adam, A. Micoulet, J. Blummel, J. Auernheimer, H. Kessler and J. P. Spatz, Lateral spacing of integrin ligands influences cell spreading and focal adhesion assembly, Eur. J. Cell Biol., 2006, 85, 219–224 CrossRef CAS PubMed.
- C. Selhuber-Unkel, T. Erdmann, M. López-García, H. Kessler, U. S. Schwarz and J. P. Spatz, Cell adhesion strength is controlled by intermolecular spacing od adhesion receptors, Biophys. J., 2010, 98, 543–551 CrossRef CAS PubMed.
- E. Mazl Chanova, P. Knotek, Y. Yang, L. Machova, V. Proks, J. Kucka, S. Popelka, O. Pop-Georgievski, A. El Haj, D. Kubies and F. Rypacek, Nano-colloid printing of functionalized PLA-b-PEO copolymers: Tailoring the surface pattern of adhesive motif and its effect on cell attachment, Physiol. Res., 2015, 64, S61–S73 Search PubMed.
- Ch. Ligoure, Surface micelles formation by adsorption of block copolymers, Macromolecules, 1991, 24, 2968–2972 CrossRef CAS.
- V. Proks, J. Jaros, O. Pop-Georgievski, J. Kucka, S. Popelka, P. Dvorak, A. Hampl and F. Rypacek, “Click & Seed” approach to the biomimetic modification of material surfaces, Macromol. Biosci., 2012, 12, 1232–1242 CrossRef CAS PubMed.
- International Organisation for Standardisation (ISO), 1996; Methods for Determination of Particle Size Distribution Part 8: Photon Correlation Spectroscopy. International Standard ISO13321.
- O. Pop-Georgievski, S. Popelka, M. Houska, D. Chvostova, V. Proks and F. Rypacek, Poly(ethylene oxide) layers grafted to dopamine-melanin anchoring layer: stability and resistance to protein adsorption, Biomacromolecules, 2011, 12, 3232–3242 CrossRef CAS PubMed.
- G. Henn, D. G. Bucknall, M. Stamm, P. Vanhoorne and R. Jerome, Chain end effects and dewetting in thin polymer films, Macromolecules, 1996, 29, 4305–4313 CrossRef CAS.
- B. Zdyrko, V. Klep and I. Luzinov, Synthesis and surface morphology of high-density poly(ethylene glycol) grafted layers, Langmuir, 2003, 19, 10179–10187 CrossRef CAS.
- B. Zdyrko, S. K. Varshney and I. Luzinov, Effect of molecular weight on synthesis and surface morphology of high-density poly(ethylene glycol) grafted layers, Langmuir, 2004, 20, 6727–6735 CrossRef CAS PubMed.
- S. Kawaguchi, G. Imai, J. Suzuki, A. Miyahara and T. Kitano, Aqueous solution properties of oligo- and poly(ethylene oxide) by static light scattering and intrinsic viscosity, Polymer, 1997, 38, 2885–2891 CrossRef CAS.
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, Z. Sedlakova, M. Houska and A. Bologna Alles, Interaction of Blood Plasma with Antifouling Surfaces, Langmuir, 2009, 25, 6328–6333 CrossRef CAS PubMed.
- N. Kasoju, D. Kubies, T. Sedlačík, O. Janoušková, J. Koubková, M. Kumorek and F. Rypacek, Polymer scaffolds with no skin-effect for tissue engineering applications fabricated by thermally induced phase separation, Biomed. Mater., 2016, 11, 015002 CrossRef PubMed.
- M. Pradny, M. Duskova-Smrckova, K. Dusek, O. Janouskova, Z. Sadakbayeva, M. Slouf and J. Michalek, Macroporous 2-hydroxyethyl methacrylate hydrogels of dual porosity for cell cultivation: morphology, swelling, permeability, and mechanical behaviour, J. Polym. Res., 2014, 21, 579 CrossRef.
- K. Emoto, Y. Nagasaki and K. Kataoka, Coating of surfaces with stabilized reactive micelles from poly(ethylene oxide)-poly(DL-lactic acid) block copolymer, Langmuir, 1999, 15, 5212–5218 CrossRef CAS.
- M. Dionzu, A. Morère, C. Roux, B. Lonetti, J.-D. Marty, C. Mingotaud, P. Joseph, D. Goudounèche, B. Payré, M. Léonetti and A.-F. Mingotaud, Comparison of methods for the fabrication and the characterization of polymer self-assemblies: what are the important parameters?, Soft Matter, 2016, 12, 2166–2176 RSC.
- G. B. Kharas, F. Sanchez-Riera and D. K. Severson, Polymers of lactic acid, in Plastics from Microbes: Microbial Synthesis of Polymers and Polymer Precursors, ed. D. P. Mobley, Carl Hanser Verlag, Munich, Germany, 1994, pp. 93–137 Search PubMed.
- G. Riess, Micellization of block copolymers, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- J. D. Andrade, L. M. Smith and D. E. Gregonis, The contact angle and interface energetics, in Surfaces and Interfacial Aspects of Biomedical Polymers: Surface Chemistry and Physics, ed. J. D. Andrade, Plenum Press, New York, USA, 1985, pp. 249–292, Biomedical polymers Search PubMed.
- B. Braun, J. R. Dorgan and S. F. Dec, Infrared Spectroscopic Determination of Lactide Concentration in Polylactide: An Improved Methodology, Macromolecules, 2006, 39, 9302–9310 CrossRef CAS.
- S. Tokumitsu, A. Liebich, S. Herrwerth, W. Eck, M. Himmelhaus and M. Grunze, Grafting of alkanethiol-terminated poly(ethylene glycol) on gold, Langmuir, 2002, 18, 8862–8870 CrossRef CAS.
- D. Horák, H. Hlídková, M. Hiraoui, M. Taverna, V. Proks, E. Mázl Chánová, C. Smadja and Z. Kučerová, Monodisperse carboxyl-functionalized poly(ethylene glycol)-coated magnetic poly(glycidyl methacrylate) microspheres: Application to the immunocapture of β-amyloid peptides, Macromol. Biosci., 2014, 14, 1590–1599 CrossRef PubMed.
- N. Hadjichristidis and S. Pispas, Designed block copolymers for ordered polymeric nanostructures, Adv. Polym. Sci., 2006, 200, 37–55 CrossRef CAS.
- T. Fujiwara, M. Miyamoto, Y. Kimura and S. Sakurai, Intriguing morphology transformation due to the macromolecular rearrangement of poly(L-lactide)-block-poly(oxyethylene): from core-shell nanoparticles to band structures via fragments of unimolecular size, Polymer, 2001, 42, 1515–1523 CrossRef CAS.
- S. Huang, S. Jiang, X. Chen and L. An, Dendritic superstructures and structure transitions of asymmetric poly(L-lactide-b-ethylene oxide) diblock copolymer thin films, Langmuir, 2009, 25, 13125–13132 CrossRef CAS PubMed.
- M. Kumorek, D. Kubies, E. Filova, M. Houska, N. Kasoju, E. Mazl Chanova, R. Matejka, M. Kryslova, L. Bacakova and F. Rypacek, Cellular responses modulated by FGF-2 adsorbed on albumin/heparin layer-by-layer assemblies, PLoS One, 2015, 10, e0125484 Search PubMed.
- L. Arcot, R. Ogaki, S. Zhang, R. L. Meyer and P. Kingshott, Optimizing the surface density of polyethylene glycol chains by grafting from binary solvent mixtures, Appl. Surf. Sci., 2015, 341, 134–141 CrossRef CAS.
- G. Wanka, H. Hoffmann and W. Ulbricht, Phase Diagrams and Aggregation Behavior of Poly (oxyethy1ene)-Poly(oxypropylene)-Poly(oxyethylene) Triblock Copolymers in Aqueous Solutions, Macromolecules, 1994, 27, 4145–4159 CrossRef CAS.
- H. M. Redhead and S. S. Davis, Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: in vitro characterisation and in vivo evaluation, J. Controlled Release, 2001, 70, 353–363 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of copolymers, AFM images of the PLLA surface, XPS spectra and (co)polymer surface composition from XPS analysis, SPR sensogram, stability test of copolymer surfaces, light microscopy micrographs of HUVEC on polymer surfaces. See DOI: 10.1039/c7bm00009j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |